TreeBreaker can infer the evolution of a discrete phenotype distribution on a phylogenetic tree, and divide the tree into segments where this distribution is constant.
For more details about how TreeBreaker works, please see the manuscript "Bayesian Inference of the Evolution of a Phenotype Distribution on a Phylogenetic Tree" by M Azim Ansari and Xavier Didelot, Genetics 2016 204:89-98 http://www.genetics.org/lookup/doi/10.1534/genetics.116.190496
TreeBreaker can be installed and used as a standalone application as described below. Alternatively, TreeBreaker can also be used as a R package, as described here.
- To compile TreeBreaker you need the GNU Scientific Library (GSL).
- The command to compile TreeBreaker is as follows (assuming that you are in the treeBreaker directory):
gcc -lgsl src/treeBreaker.c libs/knhx.c -o treeBreaker- If you need to install the GSL locally (for example if you do not have superuser privilege) you can do it as follows (assuming that you are in the treeBreaker directory):
wget ftp://ftp.gnu.org/gnu/gsl/gsl-1.16.tar.gz
tar xfz gsl-1.16.tar.gz
rm gsl-1.16.tar.gz
cd gsl-1.16 && ./configure && make
cd ../src
gcc -I ../gsl-1.16 treeBreaker.c ../libs/knhx.c -o treeBreaker ../gsl-1.16/.libs/libgsl.a -lm ./treeBreaker ../testData/testTree.newick ../testData/phenoTestFile.txt outfileIn this example there are 100 leaves in the tree and the command above should only take a few seconds to run. The output can then be visualised using the R script R/scripts/makeFigs.R which produces the following plot:
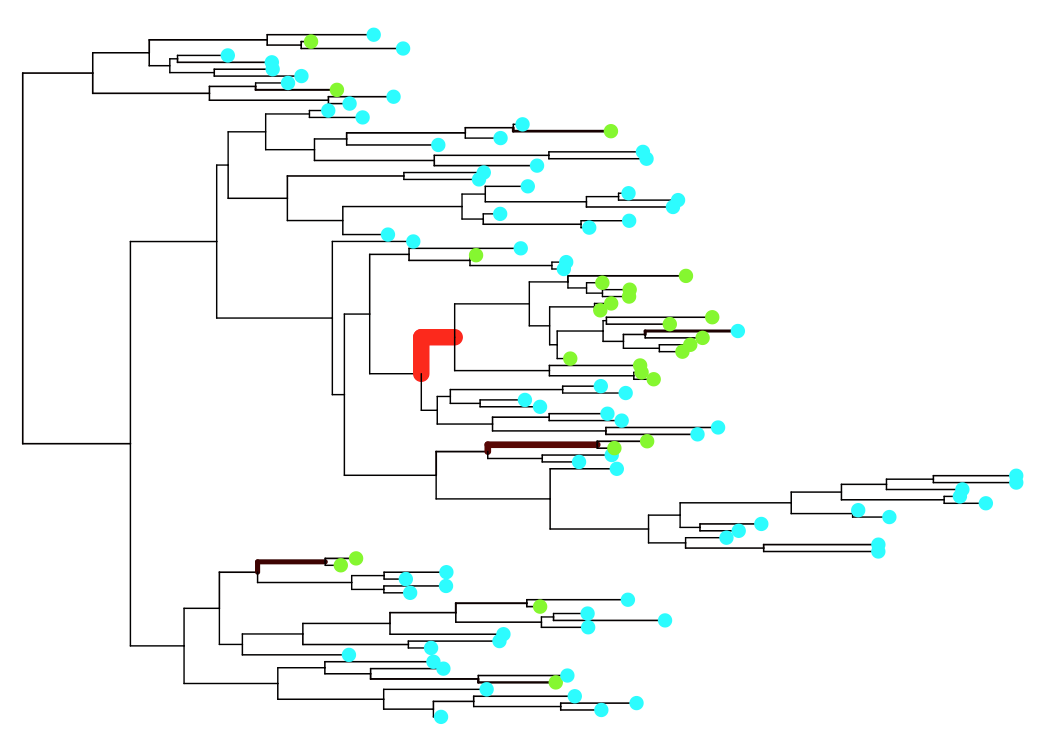
The branch thickness and colour are drawn proportional to the posterior probability of having a change point on that branch. In the above figure the clade below the thick red branch has a distinct phenotype distribution from the rest of the tree.
There are 3 mandatory input arguments which should have the following order:
- Input newick tree
- Input pheno file which has to be a tab seperated file. The first column is the name of the isolate and the second column is the phenotype. The isolate names in the pheno file have to match the isolate names in the newick file. The phenotypes have to be integers starting with 0.
- Name of output file
There are also some optional arguments, which must be indicated before the three mandatory input arguments:
-x NUM Sets the number of iterations after burn-in (default is 500000)
-y NUM Sets the number of burn-in iterations (default is 500000)
-z NUM Sets the number of iterations between samples (default is 1000)
-S NUM Sets the seed for the random number generator to NUM
-v Verbose mode
The output file contains:
- One line for each MCMC sample, with columns indicating whether branches have a changepoint on them (1) or not (0). To know which column matches which node on the tree, refer to the "index" attribute described below. The last column contains the values of lambda.
- The last line contains the newick string of the tree with comments in square brackets. Each node can have 3 attributes:
- index: shows which column in the first part of the output file matches the node. The index starts at 0.
- pheno: for leaf nodes, the phenotype that was read from the input phenotype file.
- posterior: the posterior probability of having a change point on the branch above the node.
Do contact us if you need help with using the software or you have suggestion on how to improve the R code for processing the output.
Email address: ansari dot azim @ gmail dot com dot lizard (my email address does not have dot lizard at the end of it.)