diff --git a/CONTRIBUTORS.yaml b/CONTRIBUTORS.yaml
index fe4f0afd3c299e..056c097c4d8fbb 100644
--- a/CONTRIBUTORS.yaml
+++ b/CONTRIBUTORS.yaml
@@ -86,6 +86,15 @@ abueg:
orcid: 0000-0002-6879-3954
joined: 2022-01
+adairama:
+ name: Adaikalavan Ramasamy
+ email: adai@gis.a-star.edu.sg
+ orcid: 0000-0002-7598-2892
+ linkedin: adairama
+ joined: 2024-12
+ location:
+ country: SG
+
ahmedhamidawan:
name: Ahmed Hamid Awan
email: aawan7@jhu.edu
@@ -921,6 +930,15 @@ gmauro:
- uni-freiburg
- elixir-europe
+GokceOGUZ:
+ name: Gokce Oguz
+ joined: 2024-12
+ email: gokce_oguz@gis.a-star.edu.sg
+ orcid: 0000-0003-1044-7204
+ linkedin: gokce-oguz
+ location:
+ country: SG
+
guerler:
name: Aysam Guerler
joined: 2017-09
diff --git a/_layouts/topic.html b/_layouts/topic.html
index 6c60dacafed55c..059d2b75076dc4 100644
--- a/_layouts/topic.html
+++ b/_layouts/topic.html
@@ -45,6 +45,20 @@ Not sure where to start?
{% endif %}
Material
+
+ {% if topic.toc %}
+
+
+ Jump to a section!
+
+
+
+ {% endif %}
+
You can view the tutorial materials in different languages by clicking the dropdown icon next to the slides ({% icon slides %}) and tutorial ({% icon tutorial %}) buttons below.
{% if topic.draft %}
diff --git a/bin/check-url-persistence.sh b/bin/check-url-persistence.sh
index f9c7372cbc2b8b..96be6e3436b973 100755
--- a/bin/check-url-persistence.sh
+++ b/bin/check-url-persistence.sh
@@ -24,7 +24,7 @@ cat /tmp/20*.txt | sort -u | \
grep --extended-regexp -v 'krona_?[a-z]*.html' | \
grep -v '/transcriptomics/tutorials/ref-based/faqs/rnaseq_data.html' | \
grep -v '/topics/data-management/' | \
- grep -v 'training-material/tags/' | grep -v 'data-library'| \
+ grep -v 'training-material/tags/' | grep -v 'data-library'| grep -v '/recordings/index.html' |\
sed 's|/$|/index.html|' | grep '.html$' | sort -u | sed 's|https://training.galaxyproject.org|_site|' > /tmp/gtn-files.txt
count=0
diff --git a/bin/schema-topic.yaml b/bin/schema-topic.yaml
index f3d36b8a715565..2f96b1a51290eb 100644
--- a/bin/schema-topic.yaml
+++ b/bin/schema-topic.yaml
@@ -66,6 +66,11 @@ mapping:
The image ID for an image which contains all of the tools and data for this topic.
_examples:
- quay.io/galaxy/sequence-analysis-training
+ toc:
+ type: bool
+ required: false
+ description:
+ For large topics with many subtopics, set this to true to generate a table of contents above the tutorial table to support quickly jumping to a subtopic.
subtopics:
type: seq
required: false
@@ -219,7 +224,7 @@ mapping:
type: str
description: |
The alt text for the logo (MANDATORY).
- learning_path_cta:
+ learning_path_cta:
type: str
description: |
The specific learning path you wish to reference as a call-to-action for views who aren't sure where to get started.
diff --git a/events/2022-07-08-gat.md b/events/2022-07-08-gat.md
index 6f4dd17d32a19b..b986234f4c5cca 100644
--- a/events/2022-07-08-gat.md
+++ b/events/2022-07-08-gat.md
@@ -119,7 +119,7 @@ program:
topic: admin
- name: tool-integration
topic: dev
- - name: processing-many-samples-at-once
+ - name: collections
topic: galaxy-interface
- name: upload-rules
topic: galaxy-interface
diff --git a/events/2023-04-17-gat-gent.md b/events/2023-04-17-gat-gent.md
index 145886c08bb5a5..bea47e80fb06e3 100644
--- a/events/2023-04-17-gat-gent.md
+++ b/events/2023-04-17-gat-gent.md
@@ -223,7 +223,7 @@ program:
time: "11:30 - 11:45"
- name: tool-integration
topic: dev
- - name: processing-many-samples-at-once
+ - name: collections
topic: galaxy-interface
- name: upload-rules
topic: galaxy-interface
diff --git a/faqs/galaxy/visualisations_igv.md b/faqs/galaxy/visualisations_igv.md
index 7f7119795b9a83..b0b654ed0f7231 100644
--- a/faqs/galaxy/visualisations_igv.md
+++ b/faqs/galaxy/visualisations_igv.md
@@ -4,6 +4,7 @@ area: visualisation
box_type: tip
layout: faq
contributors: [shiltemann]
+redirect_from: [/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/visualisations_igv]
---
You can send data from your Galaxy history to IGV for viewing as follows:
diff --git a/learning-pathways/admin-training.md b/learning-pathways/admin-training.md
index 46d484b6124196..436cc5c9110f3e 100644
--- a/learning-pathways/admin-training.md
+++ b/learning-pathways/admin-training.md
@@ -88,7 +88,7 @@ pathway:
topic: admin
- name: tool-integration
topic: dev
- - name: processing-many-samples-at-once
+ - name: collections
topic: galaxy-interface
- name: upload-rules
topic: galaxy-interface
diff --git a/learning-pathways/dev_tools_training.md b/learning-pathways/dev_tools_training.md
index 585ab5f3a1892a..be2c159d933192 100644
--- a/learning-pathways/dev_tools_training.md
+++ b/learning-pathways/dev_tools_training.md
@@ -23,7 +23,7 @@ pathway:
topic: admin
- name: sig_create
topic: community
-
+
- section: "Day 2: Build a batch tool"
description: This module covers getting your package on Conda, a local Galaxy instance with Planemo, write a Galaxy tool, publish it, and make it visible on a Galaxy server.
tutorials:
@@ -32,7 +32,7 @@ pathway:
- name: tools_subdomains
topic: community
- name: community-tool-table
- topic: dev
+ topic: community
- section: "Day 3: Build an interactive tool"
description: |
diff --git a/topics/community/faqs/codex.md b/topics/community/faqs/codex.md
new file mode 100644
index 00000000000000..c1c0f1fd95d1d3
--- /dev/null
+++ b/topics/community/faqs/codex.md
@@ -0,0 +1,20 @@
+---
+title: How do I add my community to the Galaxy CoDex?
+box_type: tip
+layout: faq
+contributors: [bebatut, nomadscientist]
+---
+
+You need to create a new folder in the `data/community` folder within [Galaxy Codex code source](https://github.com/galaxyproject/galaxy_codex).
+
+> Create a folder for your community
+>
+> 1. If not already done, fork the [Galaxy Codex repository](https://github.com/galaxyproject/galaxy_codex)
+> 2. Go to the `communities` folder
+> 3. Click on **Add file** in the drop-down menu at the top
+> 4. Select **Create a new file**
+> 5. Fill in the `Name of your file` field with: name of your community + `metadata/categories`
+>
+> This will create a new folder for your community and add a categories file to this folder.
+>
+{: .hands_on}
diff --git a/topics/dev/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png b/topics/community/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png
similarity index 100%
rename from topics/dev/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png
rename to topics/community/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png
diff --git a/topics/dev/tutorials/community-tool-table/images/microgalaxy_tools.png b/topics/community/tutorials/community-tool-table/images/microgalaxy_tools.png
similarity index 100%
rename from topics/dev/tutorials/community-tool-table/images/microgalaxy_tools.png
rename to topics/community/tutorials/community-tool-table/images/microgalaxy_tools.png
diff --git a/topics/dev/tutorials/community-tool-table/tutorial.md b/topics/community/tutorials/community-tool-table/tutorial.md
similarity index 83%
rename from topics/dev/tutorials/community-tool-table/tutorial.md
rename to topics/community/tutorials/community-tool-table/tutorial.md
index 7e708ec8c935e2..d54adbed6c5c94 100644
--- a/topics/dev/tutorials/community-tool-table/tutorial.md
+++ b/topics/community/tutorials/community-tool-table/tutorial.md
@@ -2,7 +2,9 @@
layout: tutorial_hands_on
title: Creation of an interactive Galaxy tools table for your community
level: Introductory
-subtopic: tooldev
+redirect_from:
+- /topics/dev/tutorials/community-tool-table/tutorial
+
questions:
- Is it possible to have an overview of all Galaxy tools for a specific scientific domain?
- How can I create a new overview for a specific Galaxy community or domain?
@@ -20,16 +22,17 @@ tags:
contributions:
authorship:
- bebatut
+ - paulzierep
---
-Galaxy offers thousands of tools. They are developed across various GitHub repositories. Furthermore, Galaxy also embraces granular implementation of software tools as sub-modules. In practice, this means that tool suites are separated into Galaxy tools, also known as wrappers, that capture their component operations. Some key examples of suites include [Mothur](https://bio.tools/mothur) and [OpenMS](https://bio.tools/openms), which translate to tens and even hundreds of Galaxy tools.
+Galaxy offers thousands of tools. They are developed across various GitHub repositories. Furthermore, Galaxy also embraces granular implementation of software tools as sub-modules. In practice, this means that tool suites are separated into Galaxy tools, also known as wrappers, that capture their component operations. Some key examples of suites include [Mothur](https://bio.tools/mothur) and [OpenMS](https://bio.tools/openms), which translate to tens and even hundreds of Galaxy tools.
-While granularity supports the composability of tools into rich domain-specific workflows, this decentralized development and sub-module architecture makes it **difficult for Galaxy users to find and reuse tools**. It may also result in Galaxy tool developers **duplicating efforts** by simultaneously wrapping the same software. This is further complicated by a lack of tool metadata, which prevents filtering for all tools in a specific research community or domain, and makes it all but impossible to employ advanced filtering with ontology terms and operations like [EDAM ontology](https://edamontology.org/page).
+While granularity supports the composability of tools into rich domain-specific workflows, this decentralized development and sub-module architecture makes it **difficult for Galaxy users to find and reuse tools**. It may also result in Galaxy tool developers **duplicating efforts** by simultaneously wrapping the same software. This is further complicated by a lack of tool metadata, which prevents filtering for all tools in a specific research community or domain, and makes it all but impossible to employ advanced filtering with ontology terms and operations like [EDAM ontology](https://edamontology.org/page).
The final challenge is also an opportunity: the global nature of Galaxy means that it is a big community. Solving the visibility of tools across this ecosystem and the potential benefits are far-reaching for global collaboration on tool and workflow development.
-To provide the research community with a comprehensive list of available Galaxy tools, [Galaxy Codex](https://github.com/galaxyproject/galaxy_codex) was developed to collect Galaxy wrappers from a list of Git repositories and automatically extract their metadata (including Conda version, [bio.tools](https://bio.tools/) identifiers, and EDAM annotations). The workflow also queries the availability of the tools and usage statistics from the three main Galaxy servers (usegalaxy.*).
+To provide the research community with a comprehensive list of available Galaxy tools, [Galaxy Codex](https://github.com/galaxyproject/galaxy_codex) was developed to collect Galaxy wrappers from a list of Git repositories and automatically extract their metadata (including Conda version, [bio.tools](https://bio.tools/) identifiers, and EDAM annotations). The workflow also queries the availability of the tools and usage statistics from the three main Galaxy servers (usegalaxy.*).

@@ -37,9 +40,9 @@ The pipeline creates an [interactive table with all tools and their metadata](ht
-The generated community-specific interactive table can be used as it and/or embedded, e.g. into the respective Galaxy Hub page or Galaxy subdomain. This table allows further filtering and searching for fine-grained tool selection.
+The generated community-specific interactive table can be used as it and/or embedded, e.g. into the respective Galaxy Hub page or Galaxy subdomain. This table allows further filtering and searching for fine-grained tool selection.
-The pipeline is **fully automated** and executes on a **weekly** basis. Any research community can apply the pipeline to create a table specific to their community.
+The pipeline is **fully automated** and executes on a **weekly** basis. Any research community can apply the pipeline to create a table specific to their community.
The aim is this tutorial is to create such table for a community.
@@ -52,21 +55,13 @@ The aim is this tutorial is to create such table for a community.
>
{: .agenda}
-# Add your community to the Galaxy Codex pipeline
+# Add your community to the Galaxy CoDex
-To create a table for a community, you first need to create a new folder in the `data/community` folder within [Galaxy Codex code source](https://github.com/galaxyproject/galaxy_codex).
+You first need to make sure that your Community is in the [Galaxy CoDex](https://github.com/galaxyproject/galaxy_codex/tree/main/communities), a central resource for Galaxy communities.
-> Create a folder for your community
->
-> 1. If not already done, fork the [Galaxy Codex repository](https://github.com/galaxyproject/galaxy_codex)
-> 2. Go to the `communities` folder
-> 3. Click on **Add file** in the drop-down menu at the top
-> 4. Select **Create a new file**
-> 5. Fill in the `Name of your file` field with: name of your community + `metadata/categories`
->
-> This will create a new folder for your community and add a categories file to this folder.
->
-{: .hands_on}
+{% snippet topics/community/faqs/codex.md %}
+
+# Add your community to the Galaxy Catalog pipeline
One of the filters for the main community table is based on the tool categories on the [Galaxy ToolShed](https://toolshed.g2.bx.psu.edu/). Only tools in the selected ToolShed categories will be added to the filtered table. As a result, it is recommended to include broad categories.
@@ -84,7 +79,7 @@ One of the filters for the main community table is based on the tool categories
>
> 4. Search on the [Galaxy ToolShed](https://toolshed.g2.bx.psu.edu/) for some of the popular tools in your community
> 5. Open the tool entries on the ToolShed, and note their categories
-> 6. Add any new categories to the `categories` file
+> 6. Add any new categories to the `categories` file
{: .hands_on}
Once you have a list of the ToolShed categories that you wish to keep, you can submit this to Galaxy Codex.
@@ -95,22 +90,22 @@ Once you have a list of the ToolShed categories that you wish to keep, you can s
> 2. Fill in the commit message with something like `Add X community`
> 3. Click on `Create a new branch for this commit and start a pull request`
> 4. Create the pull request by following the instructions
->
+>
{: .hands_on}
-The Pull Request will be reviewed. Make sure to respond to any feedback.
+The Pull Request will be reviewed. Make sure to respond to any feedback.
Once the Pull Request is merged, a table with all tool suites and a short description will be created in `communities//resources/tools_filtered_by_ts_categories.tsv`
# Review the generated table to curate tools
-The generated table will contain all the tools associated with the ToolShed categories that you selected. However, not all of these tools might be interesting for your community.
+The generated table will contain all the tools associated with the ToolShed categories that you selected. However, not all of these tools might be interesting for your community.
Galaxy Codex allows for an additional optional filter for tools, that can be defined by the community curator (maybe that is you!).
The additional filter must be stored in a file called `tools_status.tsv` located in `communities//metadata`. The file must include at least 3 columns (with a header):
1. `Suite ID`
-2. `To keep` indicating whether the tool should be included in the final table (TRUE/FALSE).
+2. `To keep` indicating whether the tool should be included in the final table (TRUE/FALSE).
3. `Deprecated` indicating whether the tool is deprecated (TRUE/FALSE).
Example of the `tools_status.tsv` file:
@@ -125,15 +120,19 @@ To generate this file, we recommend you to use the `tools_filtered_by_ts_categor
> Review tools in your community table
>
-> 1. Download the `tools_filtered_by_ts_categories.tsv` file in `communities//resources/`.
-> 2. Open `tools.tsv` with a Spreadsheet Software
-> 3. Review each line corresponding to a tool
+> 1. Download the `tools.tsv` file in `results/`.
+> 2. Open `tools.tsv` with a Spreadsheet Software.
+> 3. Review each line corresponding to a tool.
>
-> 1. Add `TRUE` to the `To keep` column if the tool should be kept, and `FALSE` if not.
-> 2. Add `TRUE` or `FALSE` also to the `Deprecated` column.
+> You can also just review some tools. Those tools that are not reviewed will have be set to `FALSE` in the `Reviewed` column of the updated table.
+> 1. Change the value in the `Reviewed` column from `FALSE` to `TRUE` (this will be done automatically if an entry of the tool in `tools_status.tsv` exists).
+> 2. Add `TRUE` to the `To keep` column if the tool should be kept, and `FALSE` if not.
+> 3. Add `TRUE` or `FALSE` also to the `Deprecated` column.
+> 4. Copy paste the `Galaxy wrapper id`, `To keep`, `Deprecated` columns in a new table (in that order).
>
-> 5. Export the new table as TSV
-> 6. Submit the TSV as `tools_status.tsv` in your `communities//metadata/` folder.
+> This can also be done using the reference function of your Spreadsheet Software.
+> 5. Export the new table as TSV (without header).
+> 6. Submit the TSV as `tools_status.tsv` in your community folder.
> 7. Wait for the Pull Request to be merged
>
{: .hands_on}
@@ -170,4 +169,3 @@ The interactive table you have created can be embedded in your community page on
# Conclusion
You now have an interactive table with Galaxy tools available for your community, and this table is embedded in a community page.
-
diff --git a/topics/contributing/faqs/github-fork-gtn.md b/topics/contributing/faqs/github-fork-gtn.md
new file mode 100644
index 00000000000000..903a5e73df0d7f
--- /dev/null
+++ b/topics/contributing/faqs/github-fork-gtn.md
@@ -0,0 +1,13 @@
+---
+title: "Forking the GTN repository"
+area: github
+box_type: tip
+layout: faq
+contributors: [hexylena, shiltemann]
+---
+
+- Go on the GitHub repository: [github.com/galaxyproject/training-material](https://github.com/galaxyproject/training-material){: width="50%"}
+- Click on the **Fork** button (top-right corner of the page)
+
+
+
diff --git a/topics/contributing/faqs/github-fork-master-main.md b/topics/contributing/faqs/github-fork-master-main.md
new file mode 100644
index 00000000000000..435b976cd64ea3
--- /dev/null
+++ b/topics/contributing/faqs/github-fork-master-main.md
@@ -0,0 +1,29 @@
+---
+title: "Updating the default branch from master to main"
+area: github
+box_type: tip
+layout: faq
+contributors: [hexylena, shiltemann]
+---
+
+If you created your fork a long time ago, the default branch on your fork may still be called **master** instead of **main**
+
+1. Point your browser to your fork of the GTN repository
+ - The url will be `https://github.com//training_material` (replacing with your GitHub username)
+
+2. Check the default branch that is shown (at top left).
+
+ 
+
+
+3. Does it say `main`?
+ - Congrats, nothing to do, **you can skip the rest of these steps**
+
+4. Does it say `master`? Then you need to update it, following the instructions below
+
+5. Go to your fork's settings (Click on the gear icon called "Settings")
+6. Find "Branches" on the left
+7. If it says master you can click on the ⇆ icon to switch branches.
+8. Select `main` (it may not be present).
+9. If it isn't present, use the pencil icon to rename `master` to `main`.
+
diff --git a/topics/contributing/faqs/github-fork-sync.md b/topics/contributing/faqs/github-fork-sync.md
new file mode 100644
index 00000000000000..e5754ed14aa2b0
--- /dev/null
+++ b/topics/contributing/faqs/github-fork-sync.md
@@ -0,0 +1,20 @@
+---
+title: "Syncing your Fork of the GTN"
+area: github
+box_type: tip
+layout: faq
+contributors: [hexylena, shiltemann]
+---
+
+Whenever you want to contribute something new to the GTN, it is important to start with an up-to-date branch. To do this, you should always update the main branch of your fork, before creating a so-called *feature branch*, a branch where you make your changes.
+
+1. Point your browser to your fork of the GTN repository
+ - The url will be `https://github.com//training_material` (replacing 'your username' with your GitHub username)
+
+2. You might see a message like "This branch is 367 commits behind galaxyproject/training-material:main." as in the screenshot below.
+
+ 
+
+3. Click the **Sync Fork** button on your fork to update it to the latest version.
+
+4. **TIP:** never work directly on your main branch, since that will make the sync process more difficult. Always create a new branch before committing your changes.
diff --git a/topics/contributing/tutorials/gitpod/tutorial.md b/topics/contributing/tutorials/gitpod/tutorial.md
index a03365a9bbb353..a43cb8e745c7e9 100644
--- a/topics/contributing/tutorials/gitpod/tutorial.md
+++ b/topics/contributing/tutorials/gitpod/tutorial.md
@@ -47,41 +47,25 @@ If you are working on your own training materials and want preview them online w
> Setting up GitPod
>
-> 1. **Create a fork** of the GTN GitHub repository
-> - Go on the GitHub repository: [github.com/galaxyproject/training-material](https://github.com/galaxyproject/training-material){: width="50%"}
-> - Click on th Fork button (top-right corner of the page)
-> 
+> 1. **Create a fork** of the [GTN GitHub repository](https://github.com/galaxyproject/training-material)
>
-> > Already have a fork of the GTN?
-> > If you already have a fork, fantastic! But a common issue is that the `main` branch gets outdated, or your fork was from before we renamed the `master` branch to `main`.
-> >
-> > - Start by browsing to your fork in GitHub
-> > - Check the default branch that is shown.
-> > - Does it say `master`? Then you need to update it, following the instructions below
-> >
-> > > changing your default branch from master to main
-> > > 1. Go to your fork's settings (Click on the gear icon called "Settings")
-> > > 2. Find "Branches" on the left
-> > > 3. If it says master you can click on the ⇆ icon to switch branches.
-> > > 4. Select `main` (it may not be present).
-> > > 5. If it isn't present, use the pencil icon to rename `master` to `main`.
-> > > 6. Now you can update it in the next step
-> > {: .tip}
-> >
-> > - Click the **Sync Fork** button on your fork to update it to the latest version
-> >
-> > 
-> {: .tip}
+> {% snippet topics/contributing/faqs/github-fork-gtn.md %}
+>
+> 2. Already have a fork of the GTN? Make sure it is up to date.
+>
+> {% snippet topics/contributing/faqs/github-fork-master-main.md %}
+>
+> {% snippet topics/contributing/faqs/github-fork-sync.md %}
>
-> 2. **Open** your browser and navigate to [gitpod.io/login](https://gitpod.io/login)
+> 3. **Open** your browser and navigate to [gitpod.io/login](https://gitpod.io/login)
> - Note: don't leave the `/login` part of the URL off, it will lead you to a different flavour of GitPod. We are using Gitpod classic
-> 3. **Log in** with GitHub
+> 4. **Log in** with GitHub
> {: width="25%"}
-> 4. Click on **Configure your own repository** under the Workspaces menu
+> 5. Click on **Configure your own repository** under the Workspaces menu
> 
-> 5. Under **Select a repository** choose your fork, e.g. `https://github.com/shiltemann/training-material`
+> 6. Under **Select a repository** choose your fork, e.g. `https://github.com/shiltemann/training-material`
> 
-> 6. Click **continue**
+> 7. Click **continue**
> - This will create an enviroment where you can make changes to the GTN and preview them
> - **Note:** It can take quite some time to start up the first time (15-30 minutes)
> - We can make future starts a lot faster using **prebuilds** (see tip box below), you can configure this now while you wait
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespace-publlish.png b/topics/contributing/tutorials/running-codespaces/images/codespace-publlish.png
new file mode 100644
index 00000000000000..019b792afaeac9
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespace-publlish.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change1.png b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change1.png
new file mode 100644
index 00000000000000..1cd8e4e4bbfd8d
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change1.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change2.png b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change2.png
new file mode 100644
index 00000000000000..582dd0b695b321
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change2.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespaces-commit-plus.png b/topics/contributing/tutorials/running-codespaces/images/codespaces-commit-plus.png
new file mode 100644
index 00000000000000..6841eba3dca3a5
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespaces-commit-plus.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/tutorial.md b/topics/contributing/tutorials/running-codespaces/tutorial.md
index f9eeb4b98620c0..9cbfae87838e02 100644
--- a/topics/contributing/tutorials/running-codespaces/tutorial.md
+++ b/topics/contributing/tutorials/running-codespaces/tutorial.md
@@ -18,12 +18,14 @@ key_points:
contributions:
authorship:
- shiltemann
+ editing:
+ - teresa-m
---
-If you are working on your own training materials and want preview them online without installing anything on your computer, you can do this using GitHub CodeSpaces! Everybody gets 60 free hours of CodeSpaces per month
+If you are working on your own training materials and want preview them online without installing anything on your computer, you can do this using GitHub CodeSpaces! Everybody gets 60 free hours of CodeSpaces per month.
>
@@ -41,17 +43,27 @@ If you are working on your own training materials and want preview them online w
> Setting up GitPod
>
-> 1. Navigate to the GTN GitHub repository, [github.com/galaxyproject/training-material](https://github.com/galaxyproject/training-material)
+> 1. **Create a fork** of the [GTN GitHub repository](https://github.com/galaxyproject/training-material)
>
-> 2. Click on the green **Code** button
+> {% snippet topics/contributing/faqs/github-fork-gtn.md %}
>
-> 3. At the top, switch to the **CodeSpaces** tab
+> 2. Already have a fork of the GTN? Make sure it is up to date.
+>
+> {% snippet topics/contributing/faqs/github-fork-master-main.md %}
+>
+> {% snippet topics/contributing/faqs/github-fork-sync.md %}
+>
+> 2. **Navigate to your fork** of the GTN
+>
+> 3. Click on the green **Code** button
+>
+> 4. At the top, switch to the **CodeSpaces** tab
> 
>
-> 4. Click on **Create codespace on main**
+> 5. Click on **Create codespace on main**
> - Note: if you switch to a specific branch in GitHub first, you can create a codespace for that branch
>
-> 5. This will setup a [Visual Studio Code](https://code.visualstudio.com/) environment for you
+> 6. This will setup a [Visual Studio Code](https://code.visualstudio.com/) environment for you
> - It may take a couple minutes to finish setting everything up
> - In this environment you can also build the GTN website to preview your changes
> - When everything is ready, you should see something like this:
@@ -178,31 +190,44 @@ When you have finished your changes, it all looks good in the preview, you want
> Comitting changes
+> Before you can commit your changes you have to create a branch. You have two options to preform this task:
+> 1. **Option 1: via the terminal**
+> - Hit ctrl+c if your preview was still running to stop it
+> - Create a new branch, commit your changes, push changes:
+>
+> ```bash
+> git checkout -b fix-title
+> git commit -m "update tutorial title" topics/introduction/tutorials/galaxy-intro-short/tutorial.md
+> git push origin fix-title
+> ```
>
-> First we commit our changes inside the codespace:
-> 1. Go to the "Source Control" icon on the left menu bar (it should have a blue dot on it)
-> 2. You should see your changed file (`tutorial.md`)
-> 
-> 3. Hover over the file name, and **click on the plus* icon* to *stage* it
-> 4. Enter a commit message (e.g. "updated tutorial title)
-> 
-> 5. Click on the green **Commit** button
+> 2. **Option 2: via the web interface**
+> - Create a new branch:
+> - On the bottom-left, click on the branch logo (probably labelled "main")
+> 
+> - Enter `fix-title` as the name for your new branch (at top of window)
+> 
+> - Choose "+ Create new branch..." from the dropdown
+> - Commit changes:
+> - On the left menu, click on the "changed files" tab
+> 
+> - You should see your changed file (`tutorial.md`)
+> - Click on the "+" icon next to the file we edited to *stage changes*
+> 
+> - Enter a commit message (top of window)
+> - Hit the checkmark icon below the massage to commit the changes
+> - Publish changes
+> - Click the cloud button at bottom left to publish your changes
+> 
>
{: .hands_on}
-Next, we will push these changes to a branch/fork. We will do this from outside of our codespace for convenience.
+Next, we will see these changes to on your branch/fork. We will do this from outside of our codespace.
> Pushing changes to GitHub
>
> 1. In your browser (outside of codespaces), navigate to the [GTN GitHub page](https://github.com/galaxyproject/training-material)
-> 2. Click on the green **Code** button again
-> 3. Click on the 3-dots menu to the right of your (randomly generated) codespace name
-> 
-> 4. Choose **Export changes to a branch**
-> - For you, it could be **Export changes to fork**
-> 
-> 5. Once it is done, click **See branch** button
-> - This will take you to the new branch
+> 2. GitHub will helpfully show you any recent branches you've pushed to your fork
> - Click the **Compare & pull request** button to create a PR for your changes
> 
{: .hands_on}
diff --git a/topics/data-science/tutorials/online-resources-protein/tutorial.md b/topics/data-science/tutorials/online-resources-protein/tutorial.md
index 289397f2745b64..9fd77c3f37e2ef 100644
--- a/topics/data-science/tutorials/online-resources-protein/tutorial.md
+++ b/topics/data-science/tutorials/online-resources-protein/tutorial.md
@@ -2,7 +2,6 @@
layout: tutorial_hands_on
title: One protein along the UniProt page
level: Introductory
-draft: true
zenodo_link: ''
questions:
- How can you search for proteins using text, gene, or protein names?
diff --git a/topics/galaxy-interface/metadata.yaml b/topics/galaxy-interface/metadata.yaml
index 5071040f57f695..b8a0078ee11261 100644
--- a/topics/galaxy-interface/metadata.yaml
+++ b/topics/galaxy-interface/metadata.yaml
@@ -5,6 +5,11 @@ title: "Using Galaxy and Managing your Data"
summary: "A collection of microtutorials explaining various features of the Galaxy user interface and manipulating data within Galaxy."
docker_image:
requirements:
+ -
+ type: "internal"
+ topic_name: introduction
+ tutorials:
+ - galaxy-intro-101
subtopics:
- id: upload
diff --git a/topics/galaxy-interface/tutorials/collections/faqs/index.md b/topics/galaxy-interface/tutorials/collections/faqs/index.md
index 9ce3fe4fce824b..c66c9e96e0565e 100644
--- a/topics/galaxy-interface/tutorials/collections/faqs/index.md
+++ b/topics/galaxy-interface/tutorials/collections/faqs/index.md
@@ -1,3 +1,5 @@
---
layout: faq-page
+redirect_from:
+- /topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/index
---
diff --git a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/visualisations_igv.md b/topics/galaxy-interface/tutorials/collections/faqs/visualisations_igv.md
similarity index 100%
rename from topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/visualisations_igv.md
rename to topics/galaxy-interface/tutorials/collections/faqs/visualisations_igv.md
diff --git a/topics/galaxy-interface/tutorials/collections/tutorial.md b/topics/galaxy-interface/tutorials/collections/tutorial.md
index 552daa1e0b61e0..aa8dab75e312f0 100644
--- a/topics/galaxy-interface/tutorials/collections/tutorial.md
+++ b/topics/galaxy-interface/tutorials/collections/tutorial.md
@@ -2,6 +2,8 @@
layout: tutorial_hands_on
redirect_from:
- /topics/galaxy-data-manipulation/tutorials/collections/tutorial
+ - /topics/galaxy-data-manipulation/tutorials/processing-many-samples-at-once/tutorial
+ - /topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial
title: "Using dataset collections"
zenodo_link: "https://doi.org/10.5281/zenodo.5119008"
@@ -46,6 +48,7 @@ recordings:
Here we will show Galaxy features designed to help with the analysis of large numbers of samples. When you have just a few samples - clicking through them is easy. But once you've got hundreds - it becomes very annoying. In Galaxy we have introduced **Dataset collections** that allow you to combine numerous datasets in a single entity that can be easily manipulated.
+
# Getting data
First, we need to upload datasets. Cut and paste the following URLs to Galaxy upload tool (see a {% icon tip %} **Tip** on how to do this [below](#tip-upload-fastqsanger-datasets-via-links)).
diff --git a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/index.md b/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/index.md
deleted file mode 100644
index 9ce3fe4fce824b..00000000000000
--- a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/index.md
+++ /dev/null
@@ -1,3 +0,0 @@
----
-layout: faq-page
----
diff --git a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.md b/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.md
deleted file mode 100644
index 5c5d9c2d64a7f1..00000000000000
--- a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.md
+++ /dev/null
@@ -1,184 +0,0 @@
----
-layout: tutorial_hands_on
-redirect_from:
- - /topics/galaxy-data-manipulation/tutorials/processing-many-samples-at-once/tutorial
-
-title: "Multisample Analysis"
-tags:
- - collections
-zenodo_link: ""
-level: Advanced
-questions:
-objectives:
-time_estimation: "1h"
-key_points:
-contributors:
- - nekrut
- - pajanne
-subtopic: manage
----
-
-Here we will show Galaxy features designed to help with the analysis of large numbers of samples. When you have just a few samples - clicking through them is easy. But once you've got hundreds - it becomes very annoying. In Galaxy we have introduced **Dataset collections** that allow you to combine numerous datasets in a single entity that can be easily manipulated.
-
-In this tutorial we assume the following:
-
-- you already have basic understanding of how Galaxy works
-- you have an account in Galaxy
-
->
->
-> In this tutorial, we will deal with:
->
-> 1. TOC
-> {:toc}
->
-{: .agenda}
-
-# Getting data
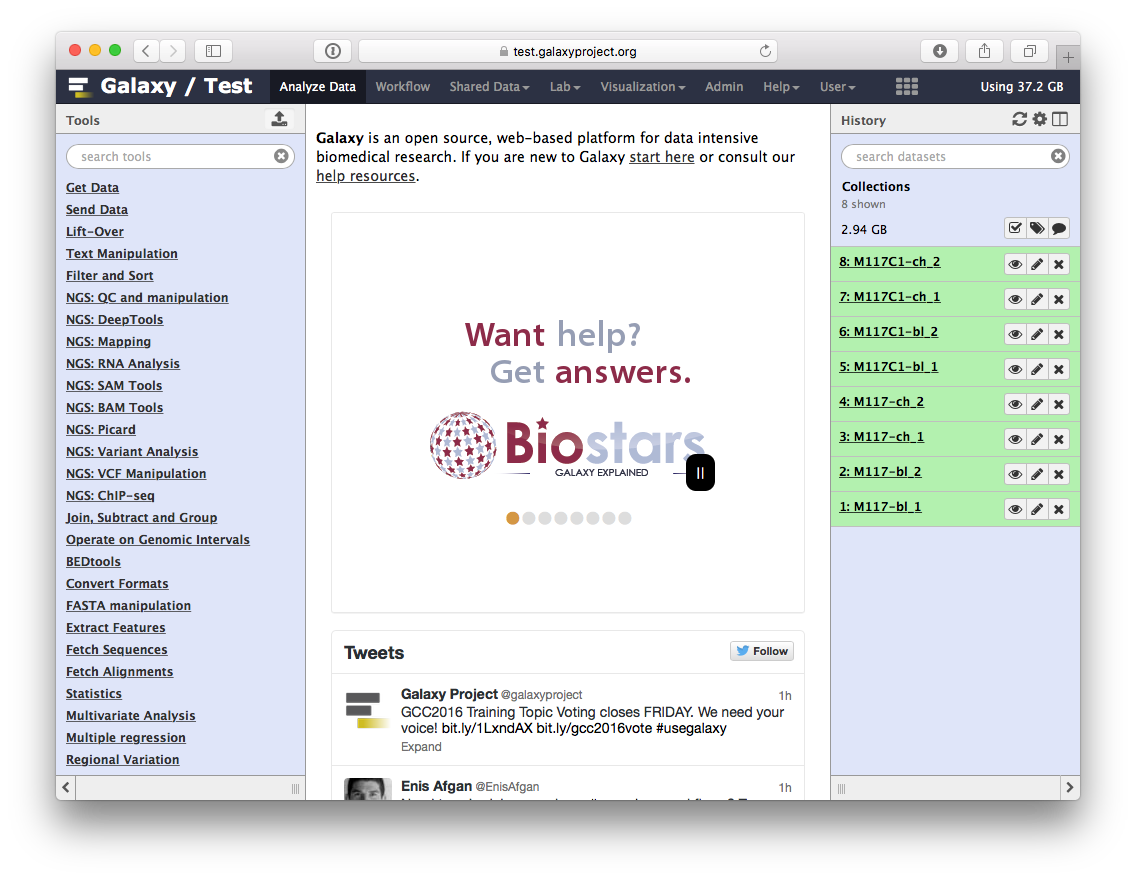
-[In this history](https://test.galaxyproject.org/u/anton/h/collections-1) are a few datasets we will be practicing with (as always with Galaxy tutorial you can upload your own data and play with it instead of the provided datasets):
-
-- `M117-bl_1` - family 117, mother, 1-st (**F**) read from **blood**
-- `M117-bl_2` - family 117, mother, 2-nd (**R**) read from **blood**
-- `M117-ch_1` - family 117, mother, 1-st (**F**) read from **cheek**
-- `M117-ch_1` - family 117, mother, 2-nd (**R**) read from **cheek**
-- `M117C1-bl_1`- family 117, child, 1-st (**F**) read from **blood**
-- `M117C1-bl_2`- family 117, child, 2-nd (**R**) read from **blood**
-- `M117C1-ch_1`- family 117, child, 1-st (**F**) read from **cheek**
-- `M117C1-ch_2`- family 117, child, 2-nd (**R**) read from **cheek**
-
-These datasets represent genomic DNA (enriched for mitochondria via a long range PCR) isolated from blood and cheek (buccal swab) of mother (`M117`) and her child (`M117C1`) that was sequenced on an Illumina miSeq machine as paired-read library (250-bp reads; see our [2014](http://www.pnas.org/content/111/43/15474.abstract) manuscript for **Methods**).
-
-# Creating a list of paired datasets
-
-If you imported [history]( https://test.galaxyproject.org/u/anton/h/collections-1) as described [above](https://github.com/nekrut/galaxy/wiki/Processing-many-samples-at-once#0-getting-data), your screen will look something like this:
-
-
-
-Now click the checkbox in  and you will see your history changing like this:
-
-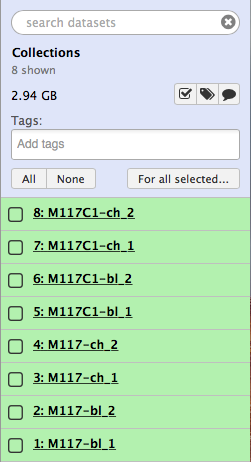
-
-Let's click `All`, which will select all datasets in the history, then click  and finally select **Build List of Dataset Pairs** from the following menu:
-
-
-
-The following wizard will appear:
-
-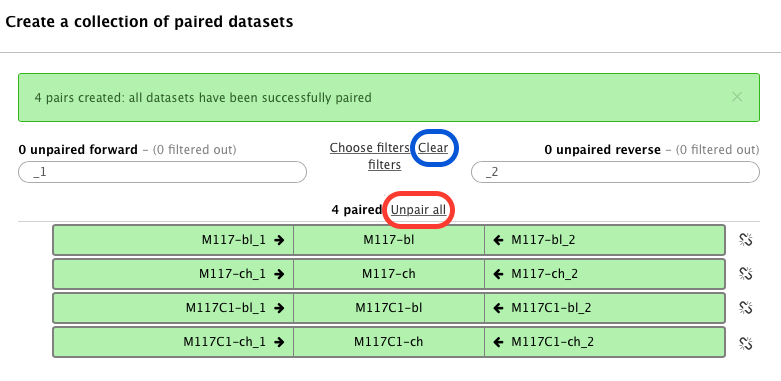
-
-In this case Galaxy automatically assigned pairs using the `_1` and `_2` endings of dataset names. Let's however pretend that this did not happen. Click on **Unpair all** (highlighted in red in the figure above) link and then on **Clear** link (highlighted in blue in the figure above). The interface will change into its virgin state:
-
-
-
-Hopefully you remember that we have paired-end data in this scenario. Datasets containing the first (forward) and the second (reverse) read are differentiated by having `_1` and `_2` in the filename. We can use this feature in dataset collection wizard to pair our datasets. Type `_1` in the left **Filter this list** text box and `_2` in the right:
-
-
-
-You will see that the dataset collection wizard will automatically filter lists on each side of the interface:
-
-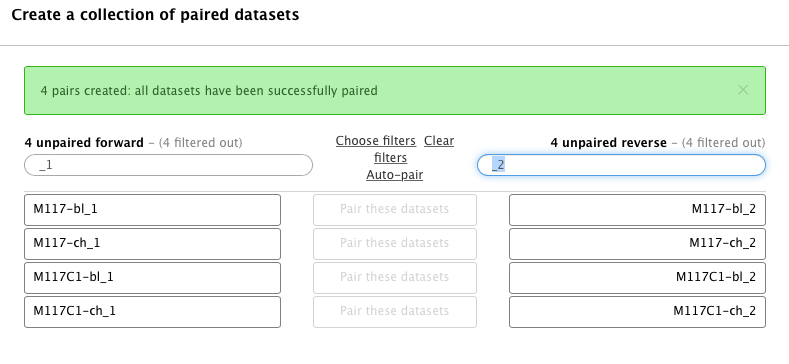
-
-Now you can either click **Auto pair** if pairs look good to you (proper combinations of datasets are listed in each line) or pair each forward/reverse group individually by pressing **Pair these datasets** button separating each pair:
-
-
-
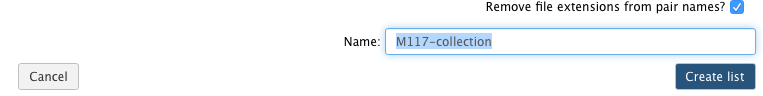
-Now it is time to name the collection:
-
-
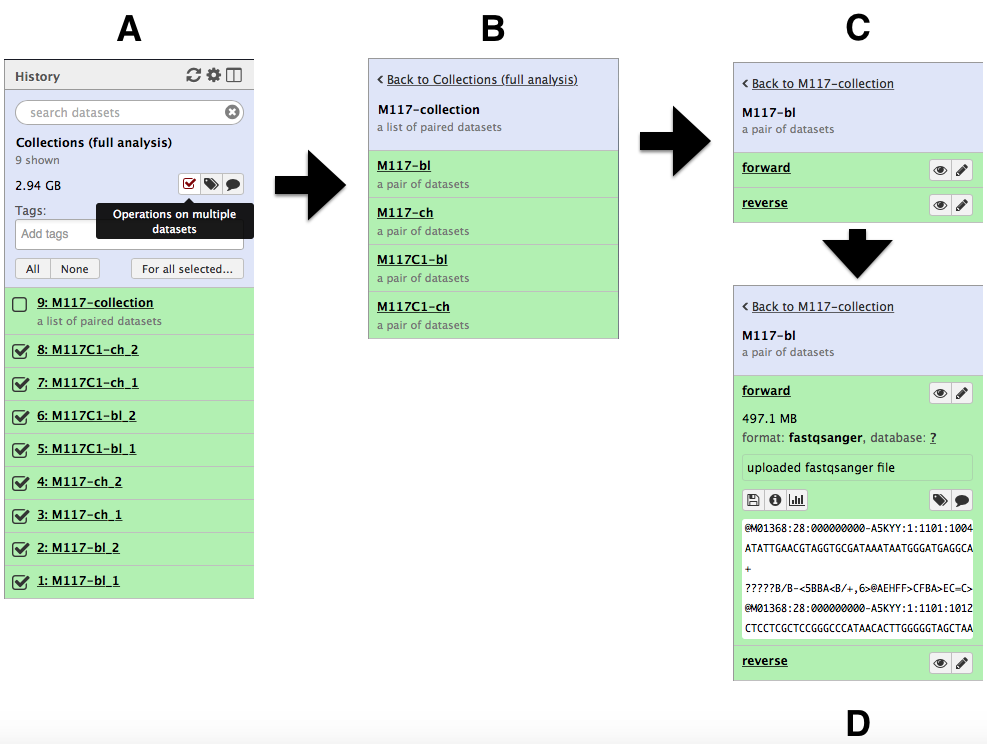
-
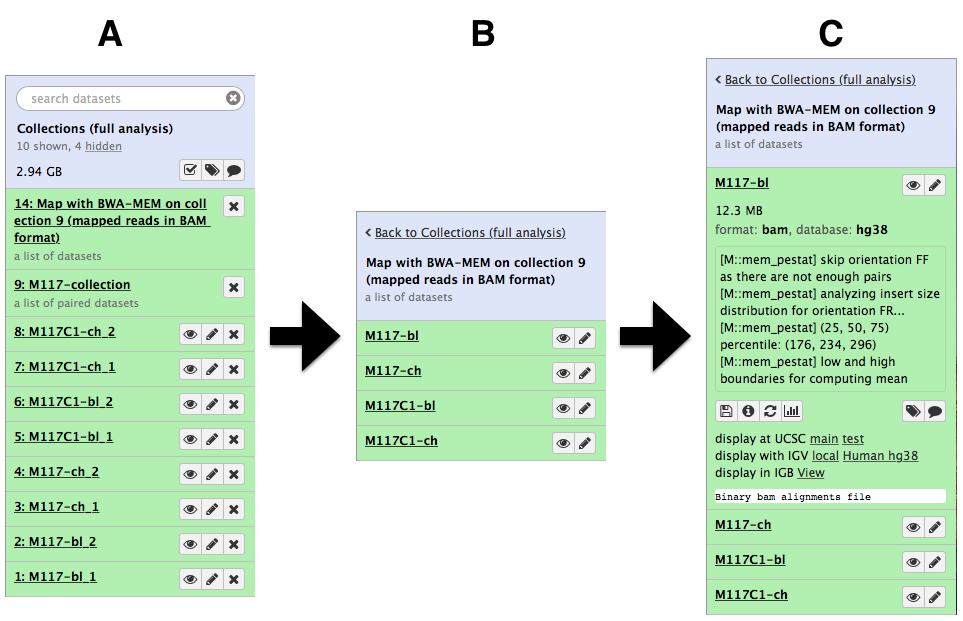
-and create the collection by clicking **Create list**. A new item will appear in the history as you can see on the panel **A** below. Clicking on collection will expand it to show four pairs it contains (panel **B**). Clicking individual pairs will expand them further to reveal **forward** and **reverse** datasets (panel **C**). Expanding these further will enable one to see individual datasets (panel **D**).
-
-
-
-# Using collections
-
-By now we see that a collection can be used to bundle a large number of items into a single history item. This means that many Galaxy tools will be able to process all datasets in a collection transparently to you. Let's try to map these datasets to human genome using `bwa-mem` mapper:
-
-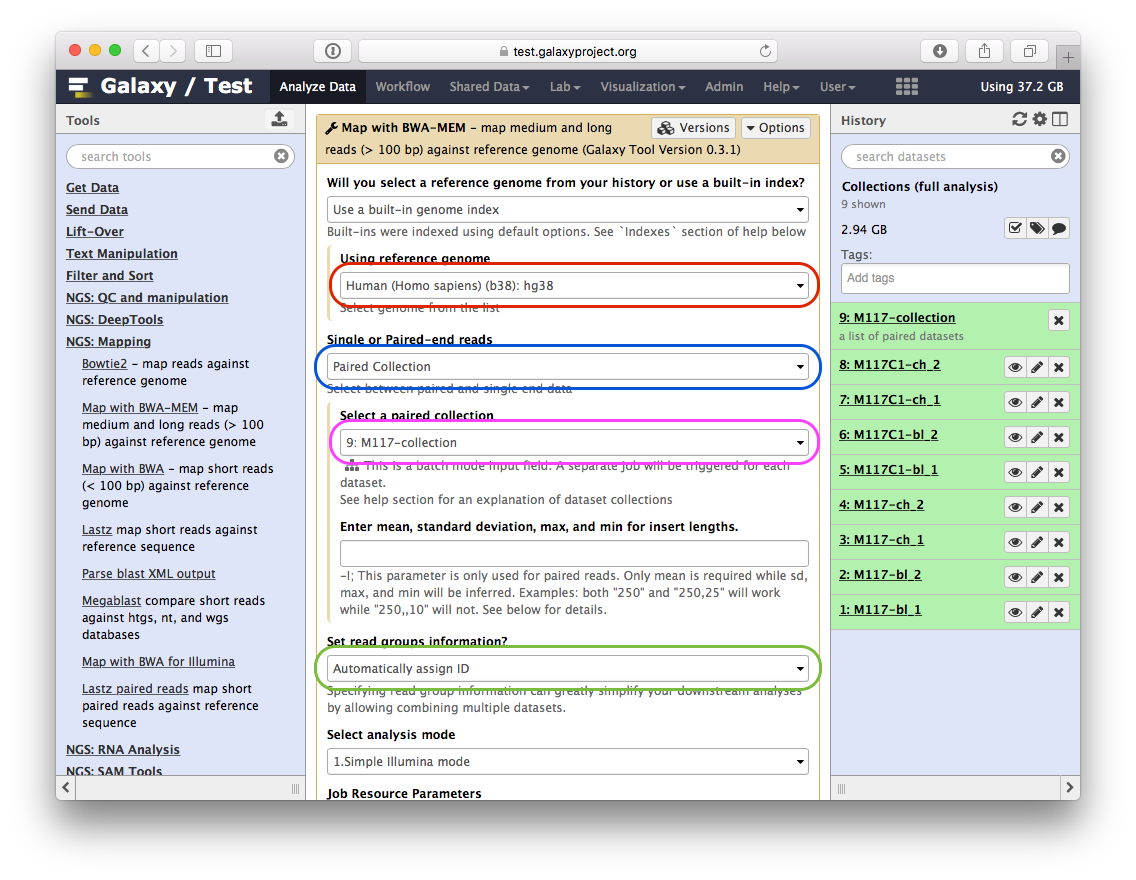
-
-Here is what you need to do:
-
-- set **Using reference genome** to `hg38` (red outline);
-- set **Single or Paired-end reads** to `Paired collection` (blue outline);
-- select `M177-collection` from **Select a paired collection** dropdown (magenta outline);
-- In **Set read groups information** select `Automatically assign ID` (green outline);
-- scroll down and click **Execute**.
-
-You will see jobs being submitted and new datasets appearing in the history. IN particular below you can see that Galaxy has started four jobs (two yellow and two gray). This is because we have eight paired datasets with each pair being processed separately by `bwa-mem`. As a result we have four `bwa-mem` runs:
-
-
-
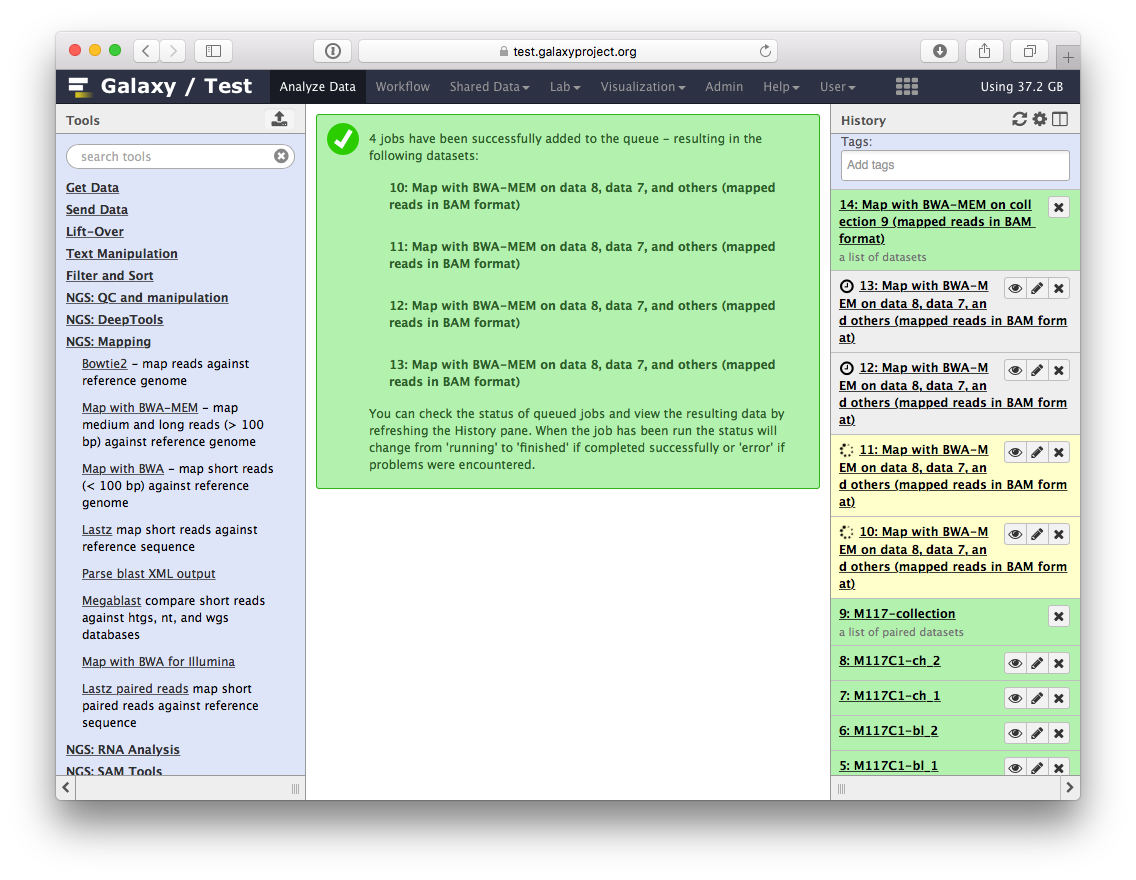
-Once these jobs are finished they will disappear from the history and all results will be represented as a new collection:
-
-
-
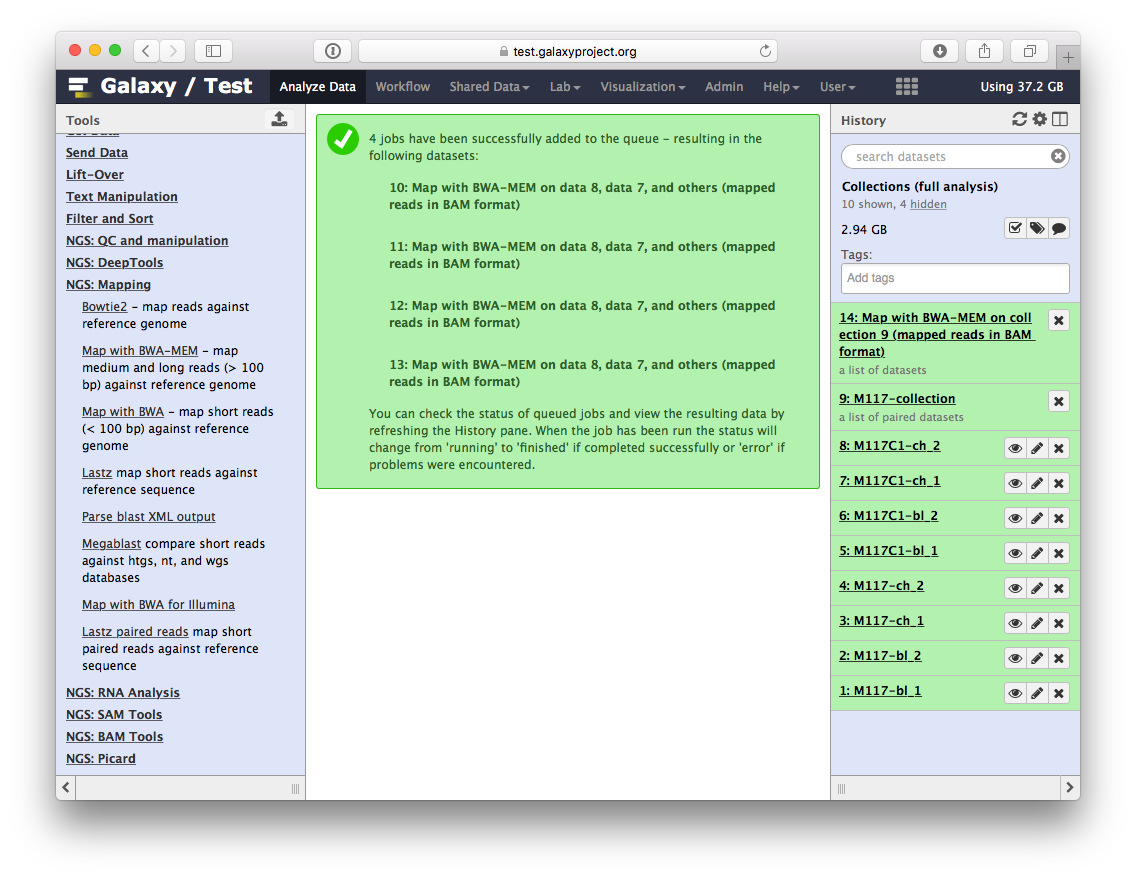
-Let's look at this collection by clicking on it (panel **A** in the figure below). You can see that now this collection is no longer paired (compared to the collection we created in the beginning of this tutorial). This is because `bwa-mem` takes forward and reverse data as input, but produces only a single BAM dataset as the output. So what we have in the result is a *list* of four dataset (BAM files; panels **B** and **C**).
-
-
-
-# Processing collection as a single entity
-
-Now that `bwa-mem` has finished and generated a collection of BAM datasets we can continue to analyze the entire collection as a single Galaxy '*item*'.
-
-## Ensuring consistency of BAM dataset
-
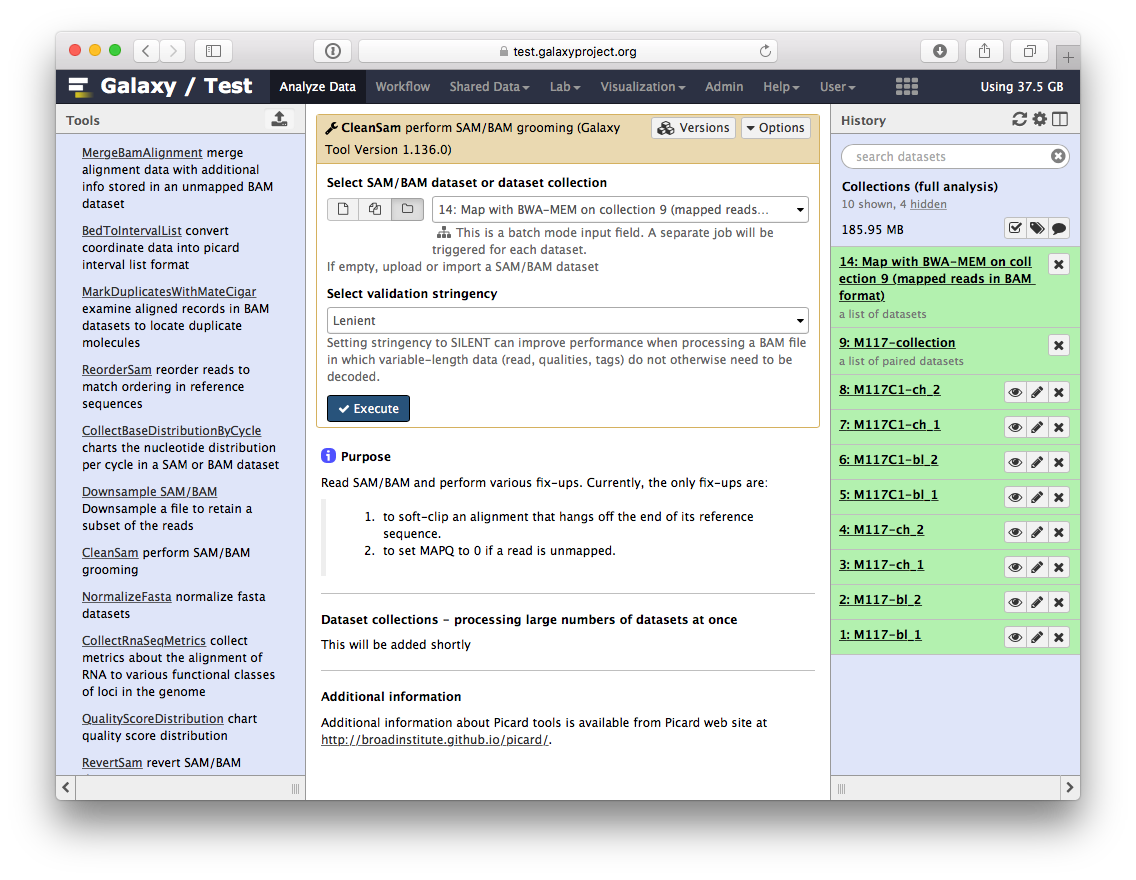
-Let's perform cleanup of our BAM files with `cleanSam` utility from the **Picard** package:
-
-
-
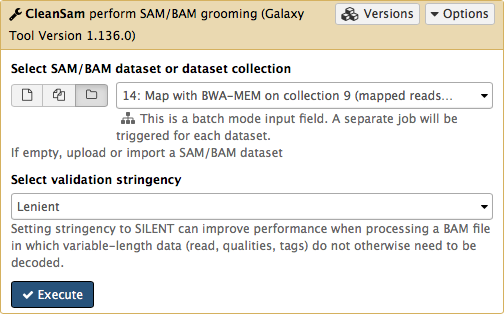
-If you look at the picture above carefully, you will see that the **Select SAM/BAM dataset or dataset collection** parameter is empty (it says `No sam or bam datasets available.`). This is because we do not have single SAM or BAM datasets in the history. Instead we have a collection. So all you need to do is to click on the **folder** () button and you will our BAM collection selected:
-
-
-
-Click **Run Tool**. As an output this tool will produce a collection contained cleaned data.
-
-## Retaining 'proper pairs'
-
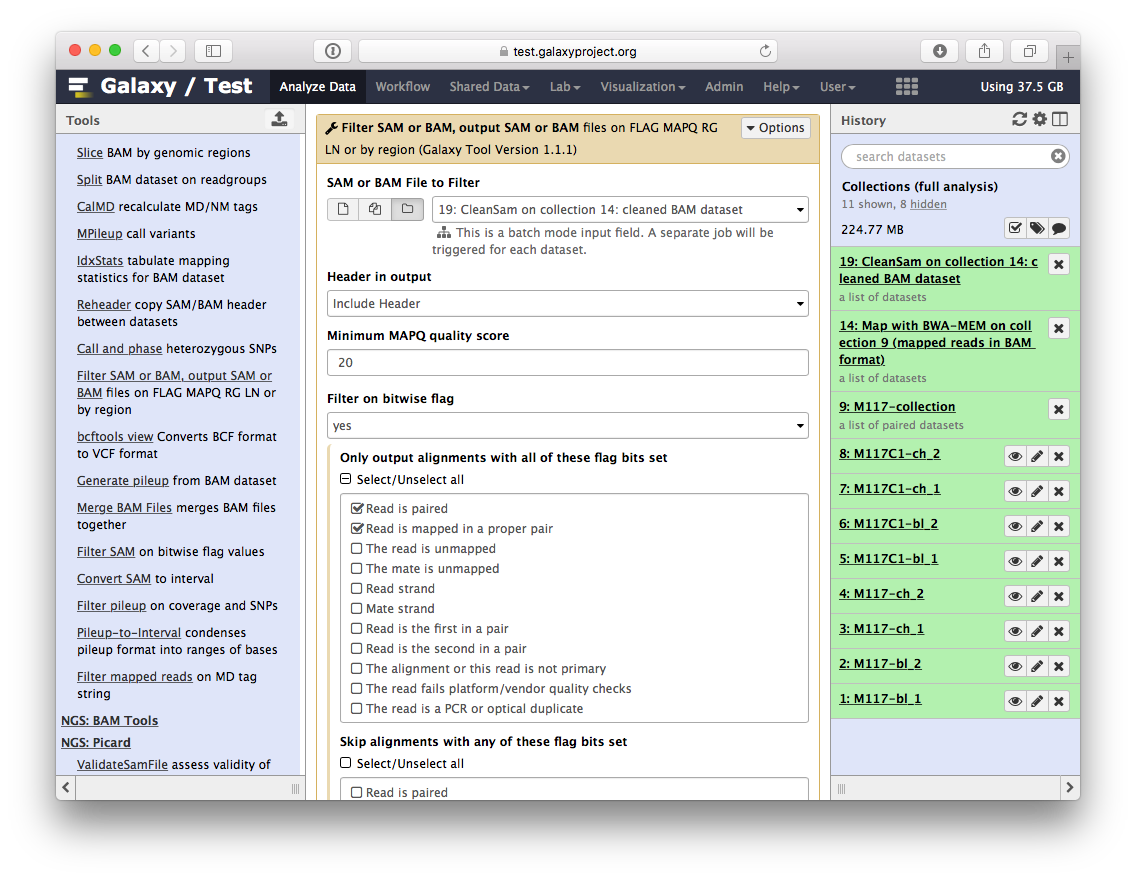
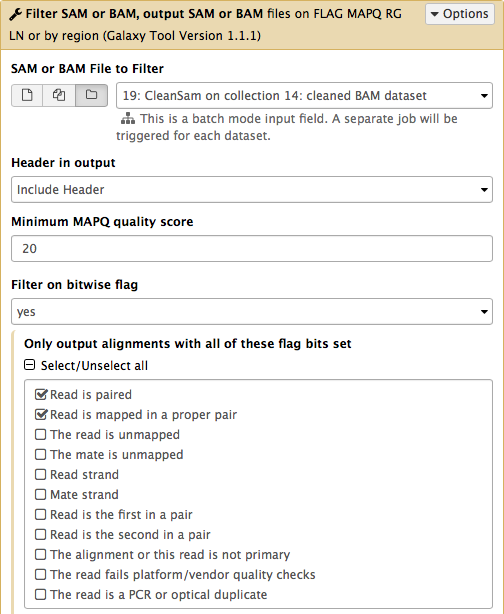
-Now let's clean the dataset further by only preserving truly paired reads (reads satisfying two requirements: (1) read is paired, and (2) it is mapped as a proper pair). For this we will use `Filter SAM or BAM` tools from **SAMTools** collection:
-
-
-
-parameters should be set as shown below. By setting mapping quality to `20` we avoid reads mapping to multiple locations and by using **Filter on bitwise flag** option we ensure that the resulting dataset will contain only properly paired reads. This operation will produce yet another collection containing now filtered datasets.
-
-
-
-## Merging collection into a single dataset
-
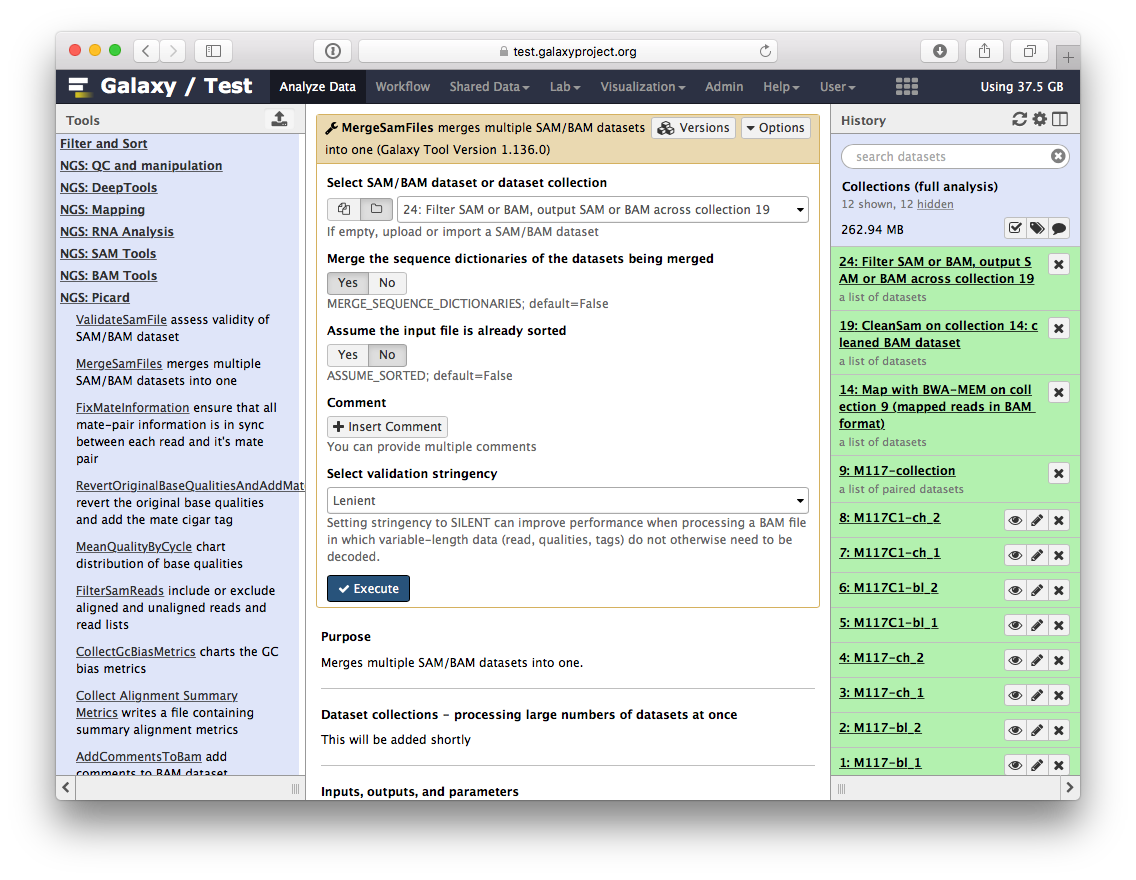
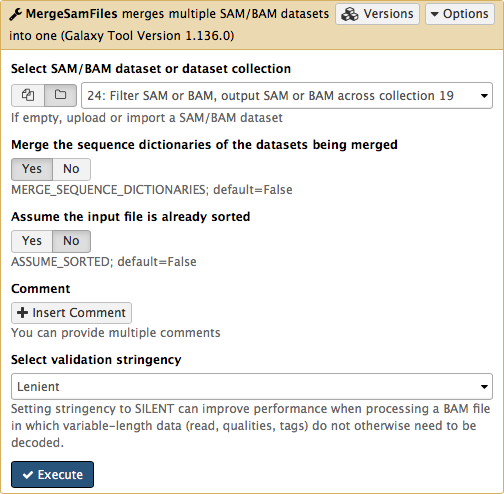
-The beauty of BAM datasets is that they can be combined in a single entity using so called *Read group* ([learn more](https://wiki.galaxyproject.org/Learn/GalaxyNGS101#Understanding_and_manipulating_SAM.2FBAM_datasets) about Read Groups on old wiki, which will be migrated here shortly). This allows to bundle reads from multiple experiments into a single dataset where read identity is maintained by labelling every sequence with *read group* tags. So let's finally reduce this collection to a single BAM dataset. For this we will use `MergeSamFiles` tool for the `Picard` suite:
-
-
-
-Here we select the collection generated by the filtering tool described above in [3.1](https://github.com/nekrut/galaxy/wiki/Processing-many-samples-at-once#31-retaining-proper-pairs):
-
-
-
-This operation will **not** generate a collection. Instead, it will generate a single BAM dataset containing mapped reads from our four samples (`M117-bl`, `M117-ch`, `M117C1-bl`, and `M117C1-ch`).
-
-# Let's look at what we've got!
-
-So we have one BAM dataset combining everything we've done so far. Let's look at the contents of this dataset using a genome browser. First, we will need to downsample the dataset to avoiding overwhelming the browser. For this we will use `Downsample SAM/BAM` tool:
-
-
-
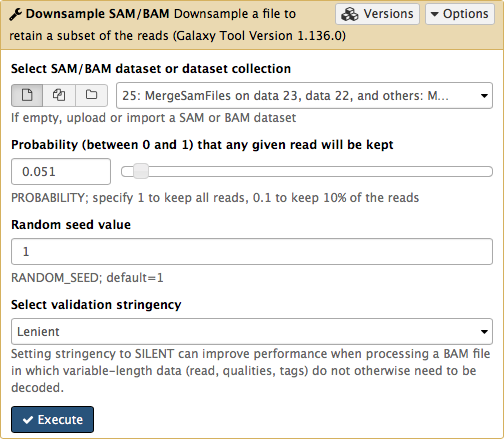
-Set **Probability (between 0 and 1) that any given read will be kept** to roughly `5%` (or `0.05`) using the slider control:
-
-
-
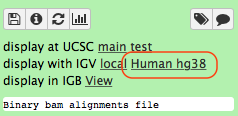
-This will generate another BAM dataset containing only 5% of the original reads and much smaller as a result. Click on this dataset and you will see links to various genome browsers:
-
-
-
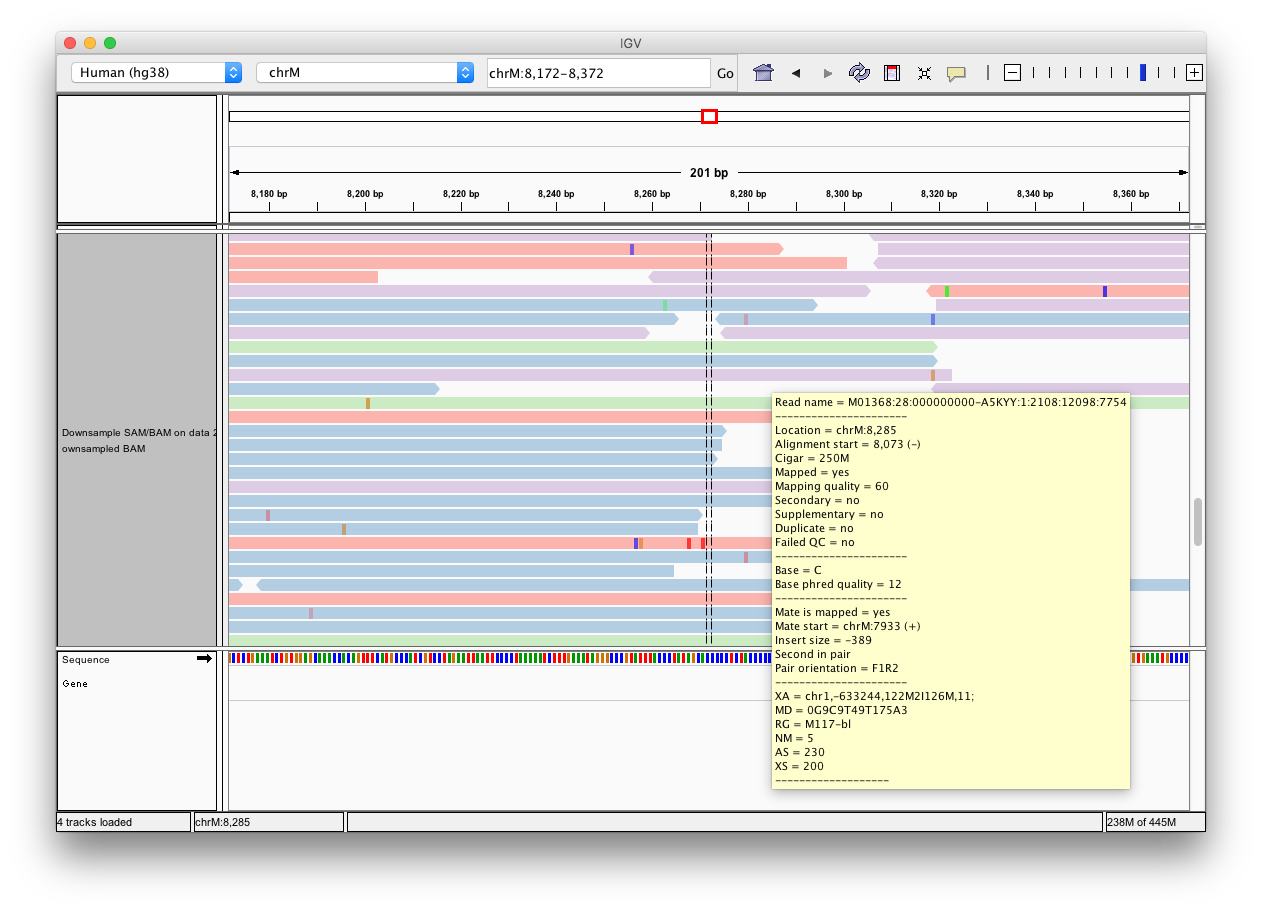
-Click the **Human hg38** link in the **display with IGV** line as highlighted above ([learn](https://wiki.galaxyproject.org/Learn/GalaxyNGS101#Visualizing_multiple_datasets_in_Integrated_Genome_Viewer_.28IGV.29) more about displaying Galaxy data in IGV with this [movie](https://vimeo.com/123442619#t=4m16s)). Below is an example generated with IGV on these data. In this screenshot reads are colored by read group (four distinct colors). A yellow inset displays additional information about a single read. One can see that this read corresponds to read group `M117-bl`.
-
-
-
-# We did not fake this:
-The two histories and the workflow described in this page are accessible directly from this page below:
-
-* History [**Collections**]( https://test.galaxyproject.org/u/anton/h/collections-1)
-* History [**Collections (full analysis)**]( https://test.galaxyproject.org/u/anton/h/collections-full-analysis)
-
-From there you can import histories to make them your own.
-
-# If things don't work...
-...you need to complain. Use [Galaxy's Help Channel](https://help.galaxyproject.org/) to do this.
diff --git a/topics/genome-annotation/tutorials/helixer/tutorial.md b/topics/genome-annotation/tutorials/helixer/tutorial.md
index d99a9ecde5f3d1..7f732f11e125fc 100644
--- a/topics/genome-annotation/tutorials/helixer/tutorial.md
+++ b/topics/genome-annotation/tutorials/helixer/tutorial.md
@@ -224,6 +224,35 @@ This gives information about the completeness of the Helixer annotation. A good
>
{: .comment}
+## Evaluation with **OMArk**
+
+[OMArk](https://github.com/DessimozLab/OMArk) is proteome quality assessment software. It provides measures of proteome completeness, characterises the consistency of all protein-coding genes with their homologues and identifies the presence of contamination by other species. OMArk is based on the OMA orthology database, from which it exploits orthology relationships, and on the OMAmer software for rapid placement of all proteins in gene families.
+
+OMArk's analysis is based on HOGs (Hierarchical Orthologous Groups), which play a central role in its assessment of the completeness and coherence of gene sets. HOGs make it possible to compare the genes of a given species with groups of orthologous genes conserved across a taxonomic clade.
+
+> OMArk on extracted protein sequences
+>
+> 1. {% tool [OMArk](toolshed.g2.bx.psu.edu/repos/iuc/omark/omark/0.3.0+galaxy2) %} with the following parameters:
+> - {% icon param-file %} *"Protein sequences"*: `gffread: pep.fa`
+> - *"OMAmer database*: select `LUCA-v2.0.0`
+> - In *"Which outputs should be generated"*: select `Detailed summary`
+>
+{: .hands_on}
+
+The OMArk tool generated an output file in .txt format containing detailed information on the assessment of the completeness, consistency and species composition of the proteome analysed. This report includes statistics on conserved genes, the proportion of duplications, missing genes and the identification of reference lineages.
+
+> What can we deduce from these results?
+>
+> - Number of conserved HOGs: OMArk has identified a set of 5622 HOGs which are thought to be conserved in the majority of species in the Mucorineae clade.
+> - 85.52% of genes are complete, so the annotation is of good quality in terms of genomic completeness.
+> - Number of proteins in the whole proteome: 19 299. Of which 62.83% are present and 30.94% of the proteome does not share sufficient similarities with known gene families.
+> - No contamination detected.
+> - The OMArk analysis is based on the Mucorineae lineage, a more recent and specific clade than that used in the BUSCO assessment, which selected the Mucorales as the reference group.
+{: .comment}
+
+
+
+
# Visualisation with a genome browser
You can visualize the annotation generated using a genomic browser like [JBrowse](https://jbrowse.org/jbrowse1.html). This browser enables you to navigate along the chromosomes of the genome and view the structure of each predicted gene.
diff --git a/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml b/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml
index 387276199fc227..94ba84e720730f 100644
--- a/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml
+++ b/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml
@@ -1,34 +1,73 @@
-- doc: Test outline for Helixer
+- doc: Test outline for Helixer Workflow
job:
- Genome:
+ Input:
class: File
+ location: https://zenodo.org/records/13890774/files/genome_masked.fa?download=1
filetype: fasta
- path: 'test-data/sequence.fasta'
outputs:
- Helixer:
- asserts:
- has_text:
- text: '##gff-version 3'
- compleasm full table:
- asserts:
- has_text:
- text: '10251at4827'
- compleasm full table busco:
- asserts:
- has_text:
- text: '10251at4827'
- compleasm miniprot:
- asserts:
- has_text:
- text: '10011at4827'
- compleasm translated protein:
- asserts:
- has_text:
- text: 'CCTCCCTCCCTCCCTCLINRLYRERLFFLGQEVDTEISNQLISLMIYLSIEKDTKDLYLFINSPGGWVISGMAIYDTMQFVRPDVQTICMGLAASIASFILVGGAITKRIAFPHAWVMIHQPASSFYEAQTGEFILEAEELLKLRETITRVYVQRTGKPIWVVSEDMERDVFMSATEAQAHGIVDLVACCTCCCTCC'
-
- BUSCO sum:
- asserts:
- has_text:
- text: '2449'
- text: '5.5.0'
+ helixer_output:
+ location: https://zenodo.org/records/13890774/files/Helixer.gff3?download=1
+ compare: sim_size
+ delta: 300000
+
+ busco_sum_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_short_summary_genome.txt?download=1
+ compare: sim_size
+ delta: 30000
+ busco_gff_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_GFF_genome.gff3?download=1
+ compare: sim_size
+ delta: 30000
+ summary_image_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_summary_image_genome.png?download=1
+ compare: sim_size
+ delta: 30000
+ busco_missing_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_missing_buscos_genome.tabular?download=1
+ compare: sim_size
+ delta: 30000
+ busco_table_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_full_table_genome.tabular?download=1
+ compare: sim_size
+ delta: 30000
+
+ gffread_pep:
+ location: https://zenodo.org/records/13890774/files/gffread_pep.fasta?download=1
+ compare: sim_size
+ delta: 30000
+
+ summary:
+ location: https://zenodo.org/records/13890774/files/genome_annotation_statistics_summary.txt?download=1
+ compare: sim_size
+ delta: 30000
+ graphs:
+ location: https://zenodo.org/records/13890774/files/genome_annotation_statistics_graphs.pdf?download=1
+ compare: sim_size
+ delta: 30000
+
+ summary_image_pep:
+ location: https://zenodo.org/records/13902305/files/Busco_pep_summary_image.png?download=1
+ compare: sim_size
+ delta: 30000
+ busco_table_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_full_table_pep.tabular?download=1
+ compare: sim_size
+ delta: 30000
+ busco_sum_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_short_summary_pep.txt?download=1
+ compare: sim_size
+ delta: 30000
+ busco_gff_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_GFF_pep.gff3?download=1
+ compare: sim_size
+ delta: 30000
+ busco_missing_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_missing_buscos_pep.tabular?download=1
+ compare: sim_size
+ delta: 30000
+
+ omark_detail_sum:
+ location: https://zenodo.org/records/13890774/files/OMArk_Detailed_summary.txt?download=1
+ compare: sim_size
+ delta: 30000
diff --git a/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga b/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga
index 42bfae7e428ec8..32bc124526514d 100644
--- a/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga
+++ b/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga
@@ -1,6 +1,105 @@
{
"a_galaxy_workflow": "true",
- "annotation": "Structural genome annotation with Helixer",
+ "annotation": "This workflow allows you to annotate a genome with Helixer and evaluate the quality of the annotation using BUSCO and Genome Annotation statistics. GFFRead is also used to predict protein sequences derived from this annotation, and BUSCO and OMArk are used to assess proteome quality. ",
+ "comments": [
+ {
+ "child_steps": [
+ 4,
+ 2
+ ],
+ "color": "lime",
+ "data": {
+ "title": "Evaluation - Genome annotation"
+ },
+ "id": 2,
+ "position": [
+ 468.3,
+ 902.5
+ ],
+ "size": [
+ 496.5,
+ 356.1
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 3
+ ],
+ "color": "orange",
+ "data": {
+ "title": "Protein prediction with Helixer annotation"
+ },
+ "id": 1,
+ "position": [
+ 628.9,
+ 255.2
+ ],
+ "size": [
+ 258,
+ 275
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 1
+ ],
+ "color": "blue",
+ "data": {
+ "title": "Annotation step"
+ },
+ "id": 0,
+ "position": [
+ 238.5,
+ 458.79999999999995
+ ],
+ "size": [
+ 240,
+ 183
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 5
+ ],
+ "color": "pink",
+ "data": {
+ "title": "Visualization"
+ },
+ "id": 4,
+ "position": [
+ 1045.3,
+ 680.0
+ ],
+ "size": [
+ 240,
+ 244.5
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 6,
+ 7
+ ],
+ "color": "turquoise",
+ "data": {
+ "title": "Evaluation - Predicted protein from annotation"
+ },
+ "id": 3,
+ "position": [
+ 1104.4,
+ 0.0
+ ],
+ "size": [
+ 312,
+ 563
+ ],
+ "type": "frame"
+ }
+ ],
"creator": [
{
"class": "Person",
@@ -10,37 +109,40 @@
],
"format-version": "0.1",
"license": "MIT",
- "name": "Training - Helixer",
+ "name": "annotation_helixer",
+ "report": {
+ "markdown": "\n# Workflow Execution Report\n\n## Workflow Inputs\n```galaxy\ninvocation_inputs()\n```\n\n## Workflow Outputs\n```galaxy\ninvocation_outputs()\n```\n\n## Workflow\n```galaxy\nworkflow_display()\n```\n"
+ },
"steps": {
"0": {
- "annotation": "Genome input (fasta format)",
+ "annotation": "Input dataset containing genomic sequences in FASTA format",
"content_id": null,
"errors": null,
"id": 0,
"input_connections": {},
"inputs": [
{
- "description": "Genome input (fasta format)",
- "name": "Genome"
+ "description": "Input dataset containing genomic sequences in FASTA format",
+ "name": "Input"
}
],
- "label": "Genome",
+ "label": "Input",
"name": "Input dataset",
"outputs": [],
"position": {
- "left": 0.0,
- "top": 393.5426496082689
+ "left": 0,
+ "top": 812.5362146249206
},
"tool_id": null,
- "tool_state": "{\"optional\": false, \"tag\": null}",
+ "tool_state": "{\"optional\": false, \"format\": [\"fasta\"], \"tag\": \"\"}",
"tool_version": null,
"type": "data_input",
- "uuid": "332fb9cd-3819-4e81-8aa5-c6cc6fbda4f7",
+ "uuid": "e267e1df-03ae-4b70-98ce-65ea177a172e",
"when": null,
"workflow_outputs": []
},
"1": {
- "annotation": "Structural annotation step",
+ "annotation": "Helixer tool for genomic annotation",
"content_id": "toolshed.g2.bx.psu.edu/repos/genouest/helixer/helixer/0.3.3+galaxy1",
"errors": null,
"id": 1,
@@ -53,7 +155,7 @@
"inputs": [
{
"description": "runtime parameter for tool Helixer",
- "name": "input"
+ "name": "input_model"
}
],
"label": "Helixer",
@@ -65,33 +167,33 @@
}
],
"position": {
- "left": 495.6559490752344,
- "top": 126.52477912175132
+ "left": 258.5333251953125,
+ "top": 498.8339807128906
},
"post_job_actions": {},
"tool_id": "toolshed.g2.bx.psu.edu/repos/genouest/helixer/helixer/0.3.3+galaxy1",
"tool_shed_repository": {
- "changeset_revision": "e3846dc36c4d",
+ "changeset_revision": "c2fc4ac35199",
"name": "helixer",
"owner": "genouest",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"input\": {\"__class__\": \"RuntimeValue\"}, \"lineages\": \"fungi\", \"option_overlap\": {\"use_overlap\": \"true\", \"__current_case__\": 0, \"overlap_offset\": null, \"overlap_core_length\": null}, \"post_processing\": {\"window_size\": \"100\", \"edge_threshold\": \"0.1\", \"peak_threshold\": \"0.8\", \"min_coding_length\": \"100\"}, \"size\": \"8\", \"species\": null, \"subsequence_length\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_state": "{\"input\": {\"__class__\": \"ConnectedValue\"}, \"input_model\": {\"__class__\": \"RuntimeValue\"}, \"lineages\": \"land_plant\", \"option_overlap\": {\"use_overlap\": \"true\", \"__current_case__\": 0, \"overlap_offset\": null, \"overlap_core_length\": null}, \"post_processing\": {\"window_size\": \"100\", \"edge_threshold\": \"0.1\", \"peak_threshold\": \"0.8\", \"min_coding_length\": \"100\"}, \"size\": \"8\", \"species\": null, \"subsequence_length\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
"tool_version": "0.3.3+galaxy1",
"type": "tool",
- "uuid": "c151bbb2-deb9-4fb6-a047-7a92ede3171a",
+ "uuid": "f60cf54d-31f2-4395-bb55-4916828cd211",
"when": null,
"workflow_outputs": [
{
- "label": "Helixer",
+ "label": "helixer_output",
"output_name": "output",
- "uuid": "9ebeb90d-1528-494c-a88d-50f28836d7c7"
+ "uuid": "fe43bcd6-5f99-4fd3-b184-2d6bfb340030"
}
]
},
"2": {
- "annotation": "Compleam is described as a faster and more accurate reimplementation of Busco.",
- "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/compleasm/compleasm/0.2.5+galaxy0",
+ "annotation": "Completeness assessment of the genome using the Busco tool",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
"errors": null,
"id": 2,
"input_connections": {
@@ -101,163 +203,135 @@
}
},
"inputs": [],
- "label": "compleasm",
- "name": "compleasm",
+ "label": "Busco on genome",
+ "name": "Busco",
"outputs": [
{
- "name": "full_table_busco",
- "type": "tsv"
+ "name": "busco_sum",
+ "type": "txt"
},
{
- "name": "full_table",
- "type": "tsv"
+ "name": "busco_table",
+ "type": "tabular"
},
{
- "name": "miniprot",
- "type": "gff3"
+ "name": "busco_missing",
+ "type": "tabular"
},
{
- "name": "translated_protein",
- "type": "fasta"
+ "name": "summary_image",
+ "type": "png"
+ },
+ {
+ "name": "busco_gff",
+ "type": "gff3"
}
],
"position": {
- "left": 677.3732598347183,
- "top": 639.0246967831238
+ "left": 744.7633406324078,
+ "top": 942.4706486763349
},
"post_job_actions": {},
- "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/compleasm/compleasm/0.2.5+galaxy0",
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
"tool_shed_repository": {
- "changeset_revision": "47f9f4d13d2c",
- "name": "compleasm",
+ "changeset_revision": "2babe6d5c561",
+ "name": "busco",
"owner": "iuc",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"__input_ext\": \"input\", \"busco_database\": \"v5\", \"chromInfo\": \"/shared/ifbstor1/galaxy/mutable-config/tool-data/shared/ucsc/chrom/?.len\", \"input\": {\"__class__\": \"ConnectedValue\"}, \"lineage_dataset\": \"mucorales_odb10\", \"mode\": \"busco\", \"outputs\": [\"full_table_busco\", \"full_table\", \"miniprot\", \"translated_protein\"], \"specified_contigs\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
- "tool_version": "0.2.5+galaxy0",
+ "tool_state": "{\"adv\": {\"evalue\": \"0.001\", \"limit\": \"3\", \"contig_break\": \"10\"}, \"busco_mode\": {\"mode\": \"geno\", \"__current_case__\": 0, \"use_augustus\": {\"use_augustus_selector\": \"augustus\", \"__current_case__\": 2, \"aug_prediction\": {\"augustus_mode\": \"no\", \"__current_case__\": 0}, \"long\": false}}, \"input\": {\"__class__\": \"ConnectedValue\"}, \"lineage\": {\"lineage_mode\": \"auto_detect\", \"__current_case__\": 0, \"auto_lineage\": \"--auto-lineage\"}, \"lineage_conditional\": {\"selector\": \"cached\", \"__current_case__\": 0, \"cached_db\": \"v5\"}, \"outputs\": [\"short_summary\", \"image\", \"gff\", \"missing\"], \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "5.7.1+galaxy0",
"type": "tool",
- "uuid": "e95adff9-488d-41af-b55c-c137ab9667cf",
+ "uuid": "c0e4cca7-0bc3-4ef2-81b2-c990b1b77d87",
"when": null,
"workflow_outputs": [
{
- "label": "compleasm miniprot",
- "output_name": "miniprot",
- "uuid": "e0d3aa0b-d18b-41af-a6fc-28a8688fe9a5"
+ "label": "busco_missing_geno",
+ "output_name": "busco_missing",
+ "uuid": "d039ef78-640f-4f7d-b449-69fac1a25130"
},
{
- "label": "compleasm full table",
- "output_name": "full_table",
- "uuid": "93b908a5-e39f-4cfb-84c3-edf4ab8b71cf"
+ "label": "busco_gff_geno",
+ "output_name": "busco_gff",
+ "uuid": "961890cc-7a33-422a-ab09-b787e3592dd1"
},
{
- "label": "compleasm full table busco",
- "output_name": "full_table_busco",
- "uuid": "7c9cb7e2-dfac-4aa1-b87d-f6a2eb7e21a5"
+ "label": "busco_sum_geno",
+ "output_name": "busco_sum",
+ "uuid": "bf09f09a-b403-4517-9a1a-acece8f36735"
},
{
- "label": "compleasm translated protein",
- "output_name": "translated_protein",
- "uuid": "9215afe6-4aad-4d43-a3a1-c2c5ab0ea1e2"
+ "label": "summary_image_geno",
+ "output_name": "summary_image",
+ "uuid": "3232c386-3c31-4989-ac76-02722ea2d79b"
+ },
+ {
+ "label": "busco_table_geno",
+ "output_name": "busco_table",
+ "uuid": "5cbbd77a-f521-4ee6-b990-a494b7671534"
}
]
},
"3": {
- "annotation": "BSUCO assesses the quality of genomic data. The tool inspects the transcription sequences of predicted genes.",
- "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.5.0+galaxy0",
+ "annotation": "Converts GFF files to other formats, such as FASTA",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/devteam/gffread/gffread/2.2.1.4+galaxy0",
"errors": null,
"id": 3,
"input_connections": {
"input": {
+ "id": 1,
+ "output_name": "output"
+ },
+ "reference_genome|genome_fasta": {
"id": 0,
"output_name": "output"
}
},
"inputs": [
{
- "description": "runtime parameter for tool Busco",
- "name": "input"
+ "description": "runtime parameter for tool gffread",
+ "name": "chr_replace"
+ },
+ {
+ "description": "runtime parameter for tool gffread",
+ "name": "reference_genome"
}
],
- "label": "BUSCO",
- "name": "Busco",
+ "label": "Gffread",
+ "name": "gffread",
"outputs": [
{
- "name": "busco_sum",
- "type": "txt"
- },
- {
- "name": "busco_table",
- "type": "tabular"
- },
- {
- "name": "busco_missing",
- "type": "tabular"
- },
- {
- "name": "summary_image",
- "type": "png"
- },
- {
- "name": "busco_gff",
- "type": "gff3"
- },
- {
- "name": "busco_miniprot",
- "type": "gff3"
+ "name": "output_pep",
+ "type": "fasta"
}
],
"position": {
- "left": 948.5586447752132,
- "top": 415.14516016871283
+ "left": 658.9081573207637,
+ "top": 316.7812237670679
},
"post_job_actions": {},
- "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.5.0+galaxy0",
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/devteam/gffread/gffread/2.2.1.4+galaxy0",
"tool_shed_repository": {
- "changeset_revision": "ea8146ee148f",
- "name": "busco",
- "owner": "iuc",
+ "changeset_revision": "3e436657dcd0",
+ "name": "gffread",
+ "owner": "devteam",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"adv\": {\"evalue\": \"0.001\", \"limit\": \"3\", \"contig_break\": \"10\"}, \"busco_mode\": {\"mode\": \"geno\", \"__current_case__\": 0, \"miniprot\": true, \"use_augustus\": {\"use_augustus_selector\": \"no\", \"__current_case__\": 0}}, \"input\": {\"__class__\": \"RuntimeValue\"}, \"lineage\": {\"lineage_mode\": \"select_lineage\", \"__current_case__\": 1, \"lineage_dataset\": \"mucorales_odb10\"}, \"lineage_conditional\": {\"selector\": \"download\", \"__current_case__\": 1}, \"outputs\": [\"short_summary\", \"missing\", \"image\", \"gff\"], \"__page__\": null, \"__rerun_remap_job_id__\": null}",
- "tool_version": "5.5.0+galaxy0",
+ "tool_state": "{\"chr_replace\": {\"__class__\": \"RuntimeValue\"}, \"decode_url\": false, \"expose\": false, \"filtering\": null, \"full_gff_attribute_preservation\": false, \"gffs\": {\"gff_fmt\": \"none\", \"__current_case__\": 0}, \"input\": {\"__class__\": \"ConnectedValue\"}, \"maxintron\": null, \"merging\": {\"merge_sel\": \"none\", \"__current_case__\": 0}, \"reference_genome\": {\"source\": \"history\", \"__current_case__\": 2, \"genome_fasta\": {\"__class__\": \"ConnectedValue\"}, \"ref_filtering\": null, \"fa_outputs\": [\"-y pep.fa\"]}, \"region\": {\"region_filter\": \"none\", \"__current_case__\": 0}, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "2.2.1.4+galaxy0",
"type": "tool",
- "uuid": "af725f3e-ee5a-485b-af11-d87a8ecf9aeb",
+ "uuid": "00d60c74-1ed5-4529-aa82-8745b50205b7",
"when": null,
"workflow_outputs": [
{
- "label": "BUSCO sum",
- "output_name": "busco_sum",
- "uuid": "703a797f-428b-4c41-984f-1283bb8eecaa"
- },
- {
- "label": "BUSCO miniprot",
- "output_name": "busco_miniprot",
- "uuid": "e3879637-613a-45cf-90a0-3eecab2e0981"
- },
- {
- "label": "BUSCO gff3",
- "output_name": "busco_gff",
- "uuid": "acbfdee0-cafd-46e8-81e9-8a1fd22ee758"
- },
- {
- "label": "BUSCO summary image",
- "output_name": "summary_image",
- "uuid": "5574f029-f31d-4d20-a847-4d1818f95707"
- },
- {
- "label": "BUSCO missing",
- "output_name": "busco_missing",
- "uuid": "b19bfb5f-901f-4568-a461-53bfc980bdcc"
- },
- {
- "label": "BUSCO table",
- "output_name": "busco_table",
- "uuid": "0ba757ff-6c1b-496b-a288-335369058923"
+ "label": "gffread_pep",
+ "output_name": "output_pep",
+ "uuid": "aa178118-cd37-495b-9e81-e2e53ebf27fd"
}
]
},
"4": {
- "annotation": "Calculate statistics from a genome annotation in GFF3 format",
+ "annotation": "Generates statistics and graphs for genome annotation",
"content_id": "toolshed.g2.bx.psu.edu/repos/iuc/jcvi_gff_stats/jcvi_gff_stats/0.8.4",
"errors": null,
"id": 4,
@@ -272,16 +346,12 @@
}
},
"inputs": [
- {
- "description": "runtime parameter for tool Genome annotation statistics",
- "name": "gff"
- },
{
"description": "runtime parameter for tool Genome annotation statistics",
"name": "ref_genome"
}
],
- "label": "Genome annotation statistics Helixer",
+ "label": "Genome annotation statistics",
"name": "Genome annotation statistics",
"outputs": [
{
@@ -294,8 +364,8 @@
}
],
"position": {
- "left": 1011.9681945236466,
- "top": 0.0
+ "left": 488.25061259116643,
+ "top": 991.5198240353345
},
"post_job_actions": {},
"tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/jcvi_gff_stats/jcvi_gff_stats/0.8.4",
@@ -305,26 +375,26 @@
"owner": "iuc",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"gff\": {\"__class__\": \"RuntimeValue\"}, \"ref_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"RuntimeValue\"}}, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_state": "{\"gff\": {\"__class__\": \"ConnectedValue\"}, \"ref_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"ConnectedValue\"}}, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
"tool_version": "0.8.4",
"type": "tool",
- "uuid": "64572fdd-797c-44e9-8662-2ae4d51528c1",
+ "uuid": "f47f89eb-23f4-4a16-b0a8-49d8e62c9f3d",
"when": null,
"workflow_outputs": [
{
- "label": "graphs helixer",
- "output_name": "graphs",
- "uuid": "a445f12b-685d-47af-b407-90fa7ea935b1"
+ "label": "summary",
+ "output_name": "summary",
+ "uuid": "fb8ed4c9-4b55-4547-880d-1916a91f8a6e"
},
{
- "label": "summary helixer",
- "output_name": "summary",
- "uuid": "8eae8a88-c73d-42dc-ad0d-62d2f1d29c6a"
+ "label": "graphs",
+ "output_name": "graphs",
+ "uuid": "4638cc23-fdb6-4e82-9cdf-c9fe38e76bd7"
}
]
},
"5": {
- "annotation": "Visualization",
+ "annotation": "JBrowse",
"content_id": "toolshed.g2.bx.psu.edu/repos/iuc/jbrowse/jbrowse/1.16.11+galaxy1",
"errors": null,
"id": 5,
@@ -353,8 +423,8 @@
}
],
"position": {
- "left": 1175.5063439719256,
- "top": 231.38681642718768
+ "left": 1065.313344724818,
+ "top": 719.9967480789329
},
"post_job_actions": {},
"tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/jbrowse/jbrowse/1.16.11+galaxy1",
@@ -364,21 +434,158 @@
"owner": "iuc",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"action\": {\"action_select\": \"create\", \"__current_case__\": 0}, \"gencode\": \"1\", \"jbgen\": {\"defaultLocation\": \"\", \"trackPadding\": \"20\", \"shareLink\": true, \"aboutDescription\": \"\", \"show_tracklist\": true, \"show_nav\": true, \"show_overview\": true, \"show_menu\": true, \"hideGenomeOptions\": false}, \"plugins\": {\"BlastView\": true, \"ComboTrackSelector\": false, \"GCContent\": false}, \"reference_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"RuntimeValue\"}}, \"standalone\": \"minimal\", \"track_groups\": [{\"__index__\": 0, \"category\": \"Annotation\", \"data_tracks\": [{\"__index__\": 0, \"data_format\": {\"data_format_select\": \"gene_calls\", \"__current_case__\": 2, \"annotation\": {\"__class__\": \"RuntimeValue\"}, \"match_part\": {\"match_part_select\": false, \"__current_case__\": 1}, \"index\": false, \"track_config\": {\"track_class\": \"NeatHTMLFeatures/View/Track/NeatFeatures\", \"__current_case__\": 3, \"html_options\": {\"topLevelFeatures\": null}}, \"jbstyle\": {\"style_classname\": \"feature\", \"style_label\": \"product,name,id\", \"style_description\": \"note,description\", \"style_height\": \"10px\", \"max_height\": \"600\"}, \"jbcolor_scale\": {\"color_score\": {\"color_score_select\": \"none\", \"__current_case__\": 0, \"color\": {\"color_select\": \"automatic\", \"__current_case__\": 0}}}, \"jb_custom_config\": {\"option\": []}, \"jbmenu\": {\"track_menu\": []}, \"track_visibility\": \"default_off\", \"override_apollo_plugins\": \"False\", \"override_apollo_drag\": \"False\"}}]}], \"uglyTestingHack\": \"\", \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_state": "{\"action\": {\"action_select\": \"create\", \"__current_case__\": 0}, \"gencode\": \"1\", \"jbgen\": {\"defaultLocation\": \"\", \"trackPadding\": \"20\", \"shareLink\": true, \"aboutDescription\": \"\", \"show_tracklist\": true, \"show_nav\": true, \"show_overview\": true, \"show_menu\": true, \"hideGenomeOptions\": false}, \"plugins\": {\"BlastView\": true, \"ComboTrackSelector\": false, \"GCContent\": false}, \"reference_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"ConnectedValue\"}}, \"standalone\": \"minimal\", \"track_groups\": [{\"__index__\": 0, \"category\": \"Annotation\", \"data_tracks\": [{\"__index__\": 0, \"data_format\": {\"data_format_select\": \"gene_calls\", \"__current_case__\": 2, \"annotation\": {\"__class__\": \"ConnectedValue\"}, \"match_part\": {\"match_part_select\": false, \"__current_case__\": 1}, \"index\": false, \"track_config\": {\"track_class\": \"NeatHTMLFeatures/View/Track/NeatFeatures\", \"__current_case__\": 3, \"html_options\": {\"topLevelFeatures\": null}}, \"jbstyle\": {\"style_classname\": \"feature\", \"style_label\": \"product,name,id\", \"style_description\": \"note,description\", \"style_height\": \"10px\", \"max_height\": \"600\"}, \"jbcolor_scale\": {\"color_score\": {\"color_score_select\": \"none\", \"__current_case__\": 0, \"color\": {\"color_select\": \"automatic\", \"__current_case__\": 0}}}, \"jb_custom_config\": {\"option\": []}, \"jbmenu\": {\"track_menu\": []}, \"track_visibility\": \"default_off\", \"override_apollo_plugins\": \"False\", \"override_apollo_drag\": \"False\"}}]}], \"uglyTestingHack\": \"\", \"__page__\": null, \"__rerun_remap_job_id__\": null}",
"tool_version": "1.16.11+galaxy1",
"type": "tool",
- "uuid": "f77404b0-6e49-4951-9553-65ef0a24499a",
+ "uuid": "04807fae-95f6-49c1-893e-76932a79cdf9",
"when": null,
"workflow_outputs": [
{
- "label": "JBrowse output",
+ "label": "output",
"output_name": "output",
- "uuid": "2bac35a7-8379-4269-9769-e0e0c541f4a3"
+ "uuid": "19976896-9df1-45e4-9c96-89e24ae6e596"
+ }
+ ]
+ },
+ "6": {
+ "annotation": "Completeness assessment of the genome using the Busco tool",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
+ "errors": null,
+ "id": 6,
+ "input_connections": {
+ "input": {
+ "id": 3,
+ "output_name": "output_pep"
+ }
+ },
+ "inputs": [],
+ "label": "Busco on protein",
+ "name": "Busco",
+ "outputs": [
+ {
+ "name": "busco_sum",
+ "type": "txt"
+ },
+ {
+ "name": "busco_table",
+ "type": "tabular"
+ },
+ {
+ "name": "busco_missing",
+ "type": "tabular"
+ },
+ {
+ "name": "summary_image",
+ "type": "png"
+ },
+ {
+ "name": "busco_gff",
+ "type": "gff3"
+ }
+ ],
+ "position": {
+ "left": 1166.6977253236494,
+ "top": 58.61198039869754
+ },
+ "post_job_actions": {},
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
+ "tool_shed_repository": {
+ "changeset_revision": "2babe6d5c561",
+ "name": "busco",
+ "owner": "iuc",
+ "tool_shed": "toolshed.g2.bx.psu.edu"
+ },
+ "tool_state": "{\"adv\": {\"evalue\": \"0.001\", \"limit\": \"3\", \"contig_break\": \"10\"}, \"busco_mode\": {\"mode\": \"prot\", \"__current_case__\": 2}, \"input\": {\"__class__\": \"ConnectedValue\"}, \"lineage\": {\"lineage_mode\": \"auto_detect\", \"__current_case__\": 0, \"auto_lineage\": \"--auto-lineage\"}, \"lineage_conditional\": {\"selector\": \"cached\", \"__current_case__\": 0, \"cached_db\": \"v5\"}, \"outputs\": [\"short_summary\", \"image\", \"gff\", \"missing\"], \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "5.7.1+galaxy0",
+ "type": "tool",
+ "uuid": "51dcc6a4-ff87-4a98-98fa-de00ce54325f",
+ "when": null,
+ "workflow_outputs": [
+ {
+ "label": "busco_gff_pep",
+ "output_name": "busco_gff",
+ "uuid": "1db166fb-10c2-4823-a80c-9f22c7c15576"
+ },
+ {
+ "label": "summary_image_pep",
+ "output_name": "summary_image",
+ "uuid": "13c6bee4-824c-4533-bc78-c99ddf0b190d"
+ },
+ {
+ "label": "busco_sum_pep",
+ "output_name": "busco_sum",
+ "uuid": "f44047d9-e713-41d9-a9f9-5543f0371d9d"
+ },
+ {
+ "label": "busco_table_pep",
+ "output_name": "busco_table",
+ "uuid": "1a113d6c-a167-432b-8200-dfb3aedc4ba1"
+ },
+ {
+ "label": "busco_missing_pep",
+ "output_name": "busco_missing",
+ "uuid": "dc2d4533-d9c2-4cb0-a144-184e90fd4e01"
+ }
+ ]
+ },
+ "7": {
+ "annotation": "OMArk",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/omark/omark/0.3.0+galaxy2",
+ "errors": null,
+ "id": 7,
+ "input_connections": {
+ "input": {
+ "id": 3,
+ "output_name": "output_pep"
+ }
+ },
+ "inputs": [
+ {
+ "description": "runtime parameter for tool OMArk",
+ "name": "input_iso"
+ }
+ ],
+ "label": "OMArk",
+ "name": "OMArk",
+ "outputs": [
+ {
+ "name": "omark_detail_sum",
+ "type": "txt"
+ },
+ {
+ "name": "omark_sum",
+ "type": "sum"
+ }
+ ],
+ "position": {
+ "left": 1167.994173976809,
+ "top": 375.00649693590475
+ },
+ "post_job_actions": {},
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/omark/omark/0.3.0+galaxy2",
+ "tool_shed_repository": {
+ "changeset_revision": "6f570ba54b41",
+ "name": "omark",
+ "owner": "iuc",
+ "tool_shed": "toolshed.g2.bx.psu.edu"
+ },
+ "tool_state": "{\"database\": \"Primates-v2.0.0.h5\", \"input\": {\"__class__\": \"ConnectedValue\"}, \"input_iso\": {\"__class__\": \"RuntimeValue\"}, \"omark_mode\": false, \"outputs\": \"detail_sum\", \"r\": null, \"t\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "0.3.0+galaxy2",
+ "type": "tool",
+ "uuid": "75e1dde7-5d60-4092-af57-cd7b065145e2",
+ "when": null,
+ "workflow_outputs": [
+ {
+ "label": "omark_detail_sum",
+ "output_name": "omark_detail_sum",
+ "uuid": "de489b9c-8808-47d4-9384-7617c33a9d34"
}
]
}
},
- "tags": [],
- "uuid": "d2fa1512-57c4-4f45-85fe-4d4bfe801f7a",
- "version": 3
-}
+ "tags": [
+ "genome-annotation"
+ ],
+ "uuid": "7a0c9f35-37a9-404e-a307-aed30a578b0c",
+ "version": 1
+}
\ No newline at end of file
diff --git a/topics/microbiome/tutorials/dada-16S/tutorial.md b/topics/microbiome/tutorials/dada-16S/tutorial.md
index c19e76fe51e55c..02f69e337fe426 100644
--- a/topics/microbiome/tutorials/dada-16S/tutorial.md
+++ b/topics/microbiome/tutorials/dada-16S/tutorial.md
@@ -130,7 +130,7 @@ To speed up analysis for this tutorial, we will use only a subset of this data.
Any analysis should get its own Galaxy history. So let's start by creating a new one:
-> Data upload
+> History creation
>
> 1. Create a new history for this analysis
>
diff --git a/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md b/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md
index 2fe9e717113a8b..15c12862c81299 100644
--- a/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md
@@ -48,7 +48,7 @@ recordings:
- katherine-d21
---
-Metaproteomics is the large-scale characterization of the entire complement of proteins expressed by microbiota. However, metaproteomics analysis of clinical samples is challenged by the presence of abundant human (host) proteins which hampers the confident detection of lower abundant microbial proteins {% cite Batut2018 %} ; [{% cite Jagtap2015 %} .
+Metaproteomics is the large-scale characterization of the entire complement of proteins expressed by microbiota. However, metaproteomics analysis of clinical samples is challenged by the presence of abundant human (host) proteins which hampers the confident detection of lower abundant microbial proteins {% cite Batut2018 %} ; {% cite Jagtap2015 %} .
To address this, we used tandem mass spectrometry (MS/MS) and bioinformatics tools on the Galaxy platform to develop a metaproteomics workflow to characterize the metaproteomes of clinical samples. This clinical metaproteomics workflow holds potential for general clinical applications such as potential secondary infections during COVID-19 infection, microbiome changes during cystic fibrosis as well as broad research questions regarding host-microbe interactions.
@@ -177,7 +177,7 @@ For this tutorial, a literature survey was conducted to obtain 118 taxonomic spe
## Merging databases to obtain a large comprehensive database for MetaNovo
Once generated, the Species UniProt database (~3.38 million sequences) will be merged with the Human SwissProt database (reviewed only; ~20.4K sequences) and contaminant (cRAP) sequences database (116 sequences) and filtered to generate the large comprehensive database (~2.59 million sequences). The large comprehensive database will be used to generate a compact database using MetaNovo, which is much more manageable.
-> Download contaminants with **Protein Database Downloader
+> Download contaminants with **Protein Database Downloader**
>
> 1. {% tool [Protein Database Downloader](toolshed.g2.bx.psu.edu/repos/galaxyp/dbbuilder/dbbuilder/0.3.4) %} with the following parameters:
> - *"Download from?"*: `cRAP (contaminants)`
diff --git a/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md b/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md
index 2c9ade8468656d..b48723580ba32b 100644
--- a/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md
@@ -487,7 +487,7 @@ MaxQuant is an MS-based proteomics platform that is capable of processing raw da
>
>
> 1. What is the Experimental Design file for MaxQuant?
-> >
+>
> >
> >
> > 1. In MaxQuant, the **Experimental Design** file is used to specify the experimental conditions, sample groups, and the relationships between different samples in a proteomics experiment. This file is a crucial component of the MaxQuant analysis process because it helps the software correctly organize and analyze the mass spectrometry data. The **Experimental Design** file typically has a ".txt" extension and is a tab-delimited text file. Here's what you might include in an Experimental Design file for MaxQuant: **Sample Names** (You specify the names of each sample in your experiment. These names should be consistent with the naming conventions used in your raw data files.), **Experimental Conditions** (You define the experimental conditions or treatment groups associated with each sample. For example, you might have control and treated groups, and you would assign the appropriate condition to each sample.), **Replicates** (You indicate the replicates for each sample, which is important for assessing the statistical significance of your results. Replicates are typically denoted by numeric values (e.g., "1," "2," "3") or by unique identifiers (e.g., "Replicate A," "Replicate B")), **Labels** (If you're using isobaric labeling methods like TMT (Tandem Mass Tag) or iTRAQ (Isobaric Tags for Relative and Absolute Quantitation), you specify the labels associated with each sample. This is important for quantification.), **Other Metadata** (You can include additional metadata relevant to your experiment, such as the biological source, time points, or any other information that helps describe the samples and experimental conditions.)
diff --git a/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md b/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md
index a75ecd0060cc01..d2c61e7ca7fd87 100644
--- a/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md
@@ -105,8 +105,8 @@ Interestingly, the PepQuery tool does not rely on searching peptides against a r
>
{: .hands_on}
-# Import Workflow
+# Import Workflow
> Running the Workflow
>
@@ -304,7 +304,8 @@ We will use the Query Tabular tool {% cite Johnson2019 %} to search the PepQuery
> > SQL Query information
> > The query input files are the list of peptides and the peptide report we obtained from MaxQuant and SGPS. The query is matching each peptide (m.pep) from the PepQuery results to the peptide reports so that each verified peptide has its protein/protein group assigned to it.
> {: .comment}
->
+{: .hands_on}
+
> Remove Header with Remove beginning
>
> 1. {% tool [Remove beginning](Remove beginning1) %} with the following parameters:
@@ -363,8 +364,8 @@ Again, we will use the Query Tabular tool to retrieve UniProt IDs (accession num
> - *"Use first line as column names"*: `Yes`
> - *"Specify Column Names (comma-separated list)"*: `pep,prot`
> ` *"SQL Query to generate tabular output"*: `SELECT distinct(prot) AS Accession
-> from t1`
-> *"include query result column headers"*: `No`
+> from t1`
+> - *"include query result column headers"*: `No`
>
>
{: .hands_on}
diff --git a/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md b/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md
index 2f9cdaab2cf8df..f21514bbb46b47 100644
--- a/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md
@@ -96,11 +96,10 @@ In this current workflow, we perform Quantification using the MaxQuant tool and
> 6. Create a dataset of the RAW files.
>
> {% snippet faqs/galaxy/datasets_add_tag.md %}
->
{: .hands_on}
-# Import Workflow
+# Import Workflow
> Running the Workflow
>
@@ -123,7 +122,7 @@ In this current workflow, we perform Quantification using the MaxQuant tool and
In the [Discovery Module](https://github.com/subinamehta/training-material/blob/main/topics/proteomics/tutorials/clinical-mp-discovery/tutorial.md), we used MaxQuant to identify peptides for verification. Now, we will again use MaxQuant to further quantify the PepQuery-verified peptides, both microbial and human. More information about quantitation using MaxQuant is available, including [Label-free data analysis](https://gxy.io/GTN:T00218) and [MaxQuant and MSstats for the analysis of TMT data](https://gxy.io/GTN:T00220).
-The outputs we are most interested in consist of the `MaxQuant Evidence file`, `MaxQuant Protein Group`s, and `MaxQuant Peptides`. The `MaxQuant Peptides` file will allow us to group them to generate a list of quantified microbial peptides.
+The outputs we are most interested in consist of the `MaxQuant Evidence file`, `MaxQuant Protein Groups`, and `MaxQuant Peptides`. The `MaxQuant Peptides` file will allow us to group them to generate a list of quantified microbial peptides.
> Quantify verified peptides (from PepQuery2)
>
diff --git a/topics/sequence-analysis/metadata.yaml b/topics/sequence-analysis/metadata.yaml
index ff22e1ba3908ea..bde7c320097d01 100644
--- a/topics/sequence-analysis/metadata.yaml
+++ b/topics/sequence-analysis/metadata.yaml
@@ -16,6 +16,11 @@ editorial_board:
- bebatut
- joachimwolff
+subtopics:
+ - id: basics
+ title: "The Basics"
+ description: "These tutorials cover basic operations common to many sequence-bases analyses"
+
references:
-
authors: "SciLifeLab"
diff --git a/topics/sequence-analysis/tutorials/mapping/tutorial.md b/topics/sequence-analysis/tutorials/mapping/tutorial.md
index 8ad00be2752e9a..b15e2a645a2dfe 100644
--- a/topics/sequence-analysis/tutorials/mapping/tutorial.md
+++ b/topics/sequence-analysis/tutorials/mapping/tutorial.md
@@ -15,6 +15,7 @@ key_points:
- Know your data!
- Mapping is not trivial
- There are many mapping algorithms, it depends on your data which one to choose
+subtopic: basics
requirements:
- type: internal
topic_name: sequence-analysis
@@ -49,9 +50,9 @@ recordings:
length: 24M
galaxy_version: 24.1.2.dev0
date: '2024-09-07'
- speakers:
+ speakers:
- DinithiRajapaksha
- captioners:
+ captioners:
- DinithiRajapaksha
bot-timestamp: 1725707919
@@ -142,7 +143,7 @@ Currently, there are over 60 different mappers, and their number is growing. In
>
> 2. Inspect the `mapping stats` file by clicking on the {% icon galaxy-eye %} (eye) icon
>
->
+>
>
{: .hands_on}
diff --git a/topics/sequence-analysis/tutorials/quality-control/tutorial.md b/topics/sequence-analysis/tutorials/quality-control/tutorial.md
index 890c3e0d9cb40f..958dd5f142350e 100644
--- a/topics/sequence-analysis/tutorials/quality-control/tutorial.md
+++ b/topics/sequence-analysis/tutorials/quality-control/tutorial.md
@@ -19,6 +19,7 @@ follow_up_training:
- mapping
time_estimation: 1H30M
level: Introductory
+subtopic: basics
key_points:
- Perform quality control on every dataset before running any other bioinformatics
analysis
diff --git a/topics/single-cell/index.md b/topics/single-cell/index.md
index a7d9d0ba06f5f6..3974bf1884dc58 100644
--- a/topics/single-cell/index.md
+++ b/topics/single-cell/index.md
@@ -7,9 +7,18 @@ topic_name: single-cell
## Want to explore analysis beyond our tutorials?
-Check out workflows shared by users like you!
-
-
+
+
+
+
Public workflows
+
+
+
+
+
News and Events
+
+
+