Eu recomendo que você trabalhe na pasta ~/dia3. Se ela não existir, crie-a com o comando: mkdir ~/dia3. Em seguida, vá para essa pasta com o comando cd ~/dia3.
As matrizes de pontos ('Dot Plot') são ferramentas exploratórias para a comparação de cadeias de texto, ou seja, sequências. Entre outras funcionalidades, elas nos permitem facilmente identificar regiões repetidas em uma sequência ao compará-la com ela mesma. Também podemos obter uma ideia bastante clara da estrutura de um gene ao comparar a sequência de sua região codificante com a sequência do locus onde ele se encontra.
Os dotplots são frequentemente usados para ilustrar comparações de genomas ou cromossomos, como exemplificado na Figura 1 deste artigo ou na Figura 2 deste outro.
Nesta seção, utilizaremos a implementação de matrizes de pontos do Instituto Suíço de Bioinformática, conhecida como Dotlet JS, para realizar comparações de pequenas sequências, conforme ilustrado na Figura:

Em seguida, estudaremos alguns exemplos provenientes da documentação do DotLet.
Faça uma comparação da sequência SLIT_DROME contra ela mesma . Copie a sequência, em seguida clique no botão 'SEQUENCE 1', faz o mesmo para o botão 'SEQUENCE 2'.
À direita dos botões onde você carregou a sequência, encontrará uma lista suspensa que permite a seleção da matriz de substituição. Logo abaixo, há outra lista suspensa que permite escolher os tamanhos da janela a serem utilizados na comparação das duas sequências.
Após o cálculo da matriz de pontos, a tela será semelhante à figura anterior. No centro da página, à esquerda, encontra-se a matriz de pontos propriamente dita, na qual padrões de linhas diagonais podem ser observados. À direita, há um histograma das pontuações das comparações dos aminoácidos.
O tamanho da janela (window size) afeta a visualização na matriz de pontos, pois determina o tamanho da área que está sendo analisada de cada vez. Janelas maiores podem mostrar uma visão mais ampla e suavizada da comparação, enquanto janelas menores podem mostrar detalhes finos e repetições mais curtas.
A linha vermelha no histograma de pontuações representa os mesmos valores das barras azuis, mas em uma escala logarítmica. Essa escala é frequentemente empregada para realçar as pontuações extremamente altas, que, de outra forma, seriam menos visíveis devido à sua baixa frequência. Isso ajuda a identificar áreas de interesse na matriz de pontos.
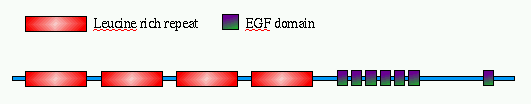
Este dotplot é da proteína SLIT de Drosophila melanogaster em relação a si mesma. Ela possui várias regiões repetidas. Na parte N-terminal (A), observamos quatro regiões repetidas, que são compostas por unidades menores repetidas (neste caso, repetições ricas em leucina). Em seguida, há outro domínio que se repete pelo menos seis vezes em um aglomerado compacto (B), com uma ocorrência adicional próxima ao C-terminus. Este último é um domínio EGF (fator de crescimento epidérmico).
A figura abaixo mostra a disposição dos domínios ao longo da sequência da proteína, conforme descrito na entrada do Swiss-Prot.
Agora carregue as sequencias MS2_HUMAN e ADAM_CROAD.
A sequência horizontal representa o antígeno de superfície celular MS2 humano, enquanto a vertical representa a adamalisina II, uma metaloprotease proveniente do veneno da serpente Crotalus adamanteus (cascavel de cauda de chocalho do leste). Ambos contêm um domínio de metaloprotease de zinco; como a imagem mostra, a adamalisina consiste apenas deste domínio, enquanto ele representa aproximadamente um quarto da proteína MS2.

Modifique os valores de Window Size para 15, 25, 35 e 45, por exemplo, e a Scoring Matrix para cada uma das matrizes BLOSUM. Como esses parâmetros afetam o dotplot?
O ANAC092 é um fator de transcrição da família NAC em Arabidopsis thaliana, identificado com o código AT5G39610.1 no genoma da planta. O gene que codifica este produto gênico tem três éxons e dois íntrons.
Carregue a sequência genômica do ANAC092 como 'SEQUENCE 1' e a sequência de cDNA como 'SEQUENCE 2' no Dotlet. Selecione a matriz Identity como Scoring Matrix e utilize vários valores de Window Size na análise. Como este parâmetro afeta a análise?

Pode identificar algum valor do parâmetro Window Size que permita visualizar sequências repetitivas no cDNA?"
O European Molecular Biology Open Software Suite EMBOSS é uma suite de software com mais de 200 aplicativos para a análise de sequências.
Nesta seção, vamos refazer os dotplots anteriores usando um aplicativo do EMBOSS. Qual aplicativo usar? Na linha de comandos, você pode usar o programa wossname para procurar aplicativos que tenham na sua descrição uma palavra-chave específica. Por exemplo, você pode procurar a palavra dotplot. Para obter mais informações sobre um aplicativo específico, você pode utilizar a página de manual man ou o comando tfm, seguido do nome do aplicativo.
Antes de utilizar os aplicativos do EMBOSS, é necessário ativar o ambiente onde o software foi instalado. Execute o seguinte comando no seu terminal. Os pacotes do EMBOSS estarão disponíveis nesse terminal. Se trocar de terminal, será necessário ativar o ambiente novamente.
conda activate emboss
Quando terminar de usar os aplicativos do EMBOSS, é importante desativar o ambiente para evitar conflitos com outros aplicativos que possam ser utilizados.
conda deactivate
Tanto o Dotlet JS quanto os programas de dotplot em EMBOSS têm algumas limitações, talvez a mais importante seja que não conseguem detectar facilmente repetições invertidas, como as que aparecem em estruturas secundárias de RNA.
Vamos usar o programa Re-Dot-Table para comparar a sequência do arquivo secondarystructure.fasta com ela mesma.
Esta sequência adquire a estrutura secundária que aparece abaixo. Nesta estrutura, as regiões que formam stems correspondem a repetições invertidas, que podem ser visualizadas como diagonais com inclinação oposta à diagonal principal no dotplot.

Para gerar o dotplot com o software Re-Dot-Table, é necessário ativar um ambiente Conda no qual o software foi previamente instalado e, em seguida, executar o aplicativo:
conda activate redotable
redotable
A seguinte figura mostra o resultado dessa comparação. Explore o efeito do tamanho da janela para gerar o dotplot.

Lembre-se de desativar seu ambiente.
conda deactivate
Além da substituição de um resíduo (aminoácido ou nucleotídeo) por outro, as sequências podem sofrer perdas ou ganhos de resíduos, ou seja, deleções ou inserções, comumente chamadas em conjunto de indels. Os dotplots não permitem incluir esse tipo de variações nas análises. Para isso, precisamos de algoritmos mais sofisticados de comparação de sequências, é aí que entram os algoritmos exatos de comparação de pares de sequências. Esses algoritmos podem ser globais ou locais.
O algoritmo para alinhamento global exato foi proposto por Needleman e Wuhsch em 1970, usa a estratégia de programação dinâmica e, para um conjunto selecionado de parâmetros, garante encontrar o melhor alinhamento possível, por isso é considerado exato. É chamado de global porque seu objetivo é encontrar o alinhamento que envolve todos os resíduos das duas sequências sendo comparadas.
O algoritmo para alinhamento local foi proposto por Smith e Waterman uma década após o desenvolvimento do algoritmo de alinhamento global. Ele também usa a estratégia de programação dinâmica e garante uma solução exata para um conjunto selecionado de parâmetros. No entanto, seu objetivo é diferente, o alinhamento produzido não precisa envolver todos os resíduos das duas sequências, mas apenas procura encontrar a subsequência mais longa comum (permitindo substituições e indels) entre as sequências de interesse.
Nesta aula prática vamos usar o programa est2genome da suite EMBOSS para alinhar uma sequencia de cDNA as uma sequencia genômica. Tenha presente que a sequencia genômica pode ter um comprimento muito maior que a sequencia de cDNA. O est2genome usa um algoritmo que combina as estrategias de alinhamento local e global. Segundo a sua propia documentacao os pasos do algoritmos são:
- Paso 1: É realizado um primeiro escaneamento Smith-Waterman para localizar a pontuação, o início e o fim do segmento com pontuação máxima (incluindo introns, é claro). Nenhuma outra informação de alinhamento é retida.
- Paso 2: Subsequências correspondentes aos segmentos de pontuação máxima são extraídas. Se o produto do comprimento dessas subsequências for menor que o parâmetro de área, os segmentos são realinhados usando o algoritmo Needleman-Wunsch, que neste caso dará o mesmo resultado que o Smith-Waterman, já que eles estão garantidos a se alinharem de ponta a ponta.
- Paso 3: Se o produto dos comprimentos exceder o limite de área, o alinhamento é dividido recursivamente, dividindo o EST ao meio e encontrando a posição no genoma que se alinha com o ponto médio do EST. O problema é então reduzido a alinhar as porções esquerda e direita das sequências separadamente e mesclar o resultado.
Por que você considera importante começar com uma varredura usando o algoritmo de Smith-Waterman?
Vamos usar a sequência genômica do ANAC092 e a sequência de cDNA para identificar o alinhamento que mostre as posições dos éxons e dos íntrons no genoma. Consulte a página de manual do est2genome e visualize o resultado do alinhamento com seu editor de texto favorito. Lembre-se de ativar o ambiente de emboss.
conda activate emboss
Vamos inspecionar a matriz BLOSUM62 que está incluída no EMBOSS. Para isso, precisamos encontrar a pasta onde o EMBOSS armazena as matrizes. Execute o seguinte comando:
embossdata EBLOSUM62
Alguma saída conterá a palavra 'exists'. No meu caso, o caminho para a matriz BLOSUM62 é /usr/share/EMBOSS/data/EBLOSUM62, e é nesse arquivo que a matriz BLOSUM62 está armazenada. Pode visualizar o conteúdo do arquivo com o comando less. Basta executar o seguinte comando:
less /usr/share/EMBOSS/data/EBLOSUM62
Isso abrirá o arquivo EBLOSUM62 no visualizador de texto "less", permitindo que você role e visualize seu conteúdo.
Responda as seguintes perguntas:
- Onde estão as maiores pontuações? Explique.
- Qual é a substituição com a maior pontuação?
- Por que as identidades não têm sempre a mesma pontuação?
Os NACs são uma família de fatores de transcrição específicos de plantas (Han et al., 2023). Neste exercício, vamos comparar uma proteína NAC de angiosperma Arabidopsis thaliana e uma proteína NAC do musgo Physcomitrium patens.
Vamos realizar um alinhamento global entre as duas proteínas. Lembre-se de que o algoritmo de alinhamento global procura o alinhamento ideal que envolva as duas sequências inteiras.
Quais programas do EMBOSS podem realizar alinhamentos globais?
wossname global
Usaremos o programa needle para realizar uma comparação global entre essas duas sequências. Verifique em timetree.org oo tempo de divergência entre as duas espécies.
Qual matriz BLOSUM considera mais adequada para comparar as duas sequências?
Execute a comparação das duas sequências, especificando a matriz selecionada, e compare os resultados usando a matriz BLOSUM90. Você pode consultar a página de manual do needle para aprender como especificar as opções:
man needle
Agora realizaremos um alinhamento local. O objetivo do alinhamento local é encontrar regiões de similaridade local, e não é necessário incluir as sequências completas. Esse tipo de alinhamento é muito útil para pesquisar bancos de dados ou quando você não tem uma ideia clara sobre a semelhança da sequência de interesse com sequências no banco de dados. Usaremos o programa water para realizar uma comparação local entre essas duas sequências. Você pode consultar a página de manual do water para ajudar na selecao de opções:
man water
Quão significativos são esses alinhamentos? Tente gerar uma sequência aleatória a partir de ANAC092 e refaça os alinhamentos exatos. Como a pontuação do alinhamento muda?
Os alinhamentos exatos representam um grande desafio computacional em termos de recursos necessários. Quando estamos buscando uma sequência semelhante a um alvo em um banco de dados que contém milhões de sequências, muitas vezes é necessário relaxar os critérios de busca para obter respostas rápidas e satisfatórias, mesmo que não sejam a resposta perfeita (exata). É aqui que ferramentas como o Basic Local Alignment Search Tool (Altschul et al., 1990), podem e devem ser empregadas. É importante destacar que o BLAST é uma ferramenta projetada para realizar alinhamentos locais, permitindo encontrar regiões similares em sequências, em vez de buscar por correspondências globais.
O BLAST possui uma interface gráfica (Grafical User Interface ou GUI) muito boa para buscar sequências no banco de dados da NCBI.
Utilize a sequência encontrada no arquivo files/unknown_nuc.fasta para realizar uma pesquisa BLAST, no site (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Esta sequencia corresponde a um trascrito de A. thaliana identificado num experimento. Realize a busca usando o programa blastn, i.e., "Nucleotide - Nucleotide" no genoma completo de A. thaliana. Quais opções você deve selecionar para restringir sua pesquisa aos cromossomos de Arabidopsis thaliana? Uma vez que o BLAST realiza a pesquisa usando alinhamentos locais, esse resultado fornecerá apenas uma ideia muito preliminar da localização do transcrito no genoma. No entanto, você pode usar essas informações para refinar a predição do locus do transcrito usando o est2genome do EMBOSS ou o Splign do NCBI. Quais opções você selecionou para realizar a pesquisa no BLAST? Por quê? Descreva os resultados da pesquisa.

Os resultados dessa pesquisa nos permitem concluir que o locus do transcrito está no cromossomo número 5 de A. thaliana. Quais são as coordenadas aproximadas no cromossomo? Existem exons? Explique sua resposta. Vamos usar esse resultado como entrada para o est2genome. Primeiro, extraia da sequência do cromossomo 5 a região detectada pelo BLAST, adicionando 5000 pb a montante e a jusante. Como você pode fazer isso? Use o est2genome para refinar a predição do locus. Quais vantagens o est2genome oferece em comparação com um simples BLAST?
No entanto, executar o BLAST através da linha de comando tem muitos benefícios:
- É muito mais fácil executar várias consultas do BLAST usando a linha de comando do que a GUI.
- A execução do BLAST com a linha de comando é reprodutível e pode ser documentada em um script.
- Os resultados podem ser salvos em um formato legível por máquina que pode ser analisado posteriormente.
- Você pode criar seus próprios bancos de dados para pesquisa em vez de usar os bancos de dados pré-construídos do NCBI.
- Isso permite a automação das consultas.
- Isso permite que você use um servidor remoto para executar as consultas do BLAST.
Ao utilizar o BLAST, é comum termos uma sequência de interesse, conhecida como query, que será comparada com sequências de um banco de dados, chamadas de subject. Neste exercício, iremos comparar as sequências de proteínas do camundongo (Mus musculus) com as sequências de proteínas do zebrafish (Danio rerio). O objetivo principal é encontrar, para cada proteína do camundongo, a proteína mais semelhante no zebrafish.
Vamos baixar os arquivos com as sequências das proteínas no formato FASTA:
curl -o mouse.1.protein.faa.gz -L https://osf.io/v6j9x/download
curl -o zebrafish.1.protein.faa.gz -L https://osf.io/68mgf/download
Descompacte-os:
gunzip *.faa.gz
E vamos dar uma olhada nas primeiras sequências no arquivo:
head mouse.1.protein.faa
Essas são sequências de proteínas no formato FASTA. O formato FASTA é algo que muitos de vocês provavelmente já viram de uma forma ou de outra - é bastante comum. É um arquivo de texto que contém registros; cada registro começa com uma linha que começa com um '>' e, em seguida, contém uma ou mais linhas de texto de sequência.
Vamos pegar essas duas primeiras sequências e salvá-las em um arquivo. Faremos isso usando a redireção de saída com o '>' que diz "pegue toda a saída e coloque-a neste arquivo aqui.". Vamos utilizar o comando head para visualizar as X primeiras linhas que contêm as duas primeiras sequências? Como podemos encontrar o valor de X?
Já estamos familiarizados com o comando grep, que nos ajuda a encontrar padrões em arquivos de texto. Neste caso, podemos procurar pelo padrão '>'. Consultando a documentação do comando grep, encontraremos a opção -n, que, além de mostrar a linha que contém o padrão, também exibirá o número da linha no arquivo. Portanto, vamos usar o grep para exibir as três primeiras linhas que contêm o sinal '>' e seus números de linha correspondentes no arquivo:"
grep -n ">" mouse.1.protein.faa|head -n3
Com esse resultado, agora sabemos que a terceira sequência começa na linha 12, ou seja, a segunda sequência termina na linha 11. O valor de X é 11, agora podemos extrair as duas primeiras sequencias do arquivo usando o comando head:
head -n 11 mouse.1.protein.faa > mm-first.faa
Agora, por exemplo, você pode usar cat mm-first.faa para ver o conteúdo desse arquivo (ou less mm-first.faa). DICA: se você tentar less mm-first.faa, precisará sair pressionando a tecla 'q' no teclado.
Agora vamos fazer um BLAST com essas duas sequências em relação a todo o conjunto de dados de proteínas do zebrafish. Primeiro, precisamos informar ao BLAST que as sequências do zebrafish são (a) um banco de dados e (b) um banco de dados de proteínas. Isso é feito chamando o 'makeblastdb'. Observe que você precisará primeiro ativar seu ambiente Conda que possui o BLAST instalado.
conda activate blast
makeblastdb -in zebrafish.1.protein.faa -dbtype prot
Em seguida, chamamos o BLAST para fazer a pesquisa:
blastp -query mm-first.faa -db zebrafish.1.protein.faa
Isso não deve tardar muito, mas você receberá muita saída na tela do computador!! Para salvá-lo em um arquivo em vez de vê-lo na tela, peça ao BLAST para salvar a saída em um arquivo que chamaremos de mm-first.x.zebrafish.txt:
blastp -num_threads 5 -query mm-first.faa -db zebrafish.1.protein.faa -out mm-first.x.zebrafish.txt
Agora, você pode 'navegar' por este arquivo à vontade digitando:
less mm-first.x.zebrafish.txt
(Tecle espaço para mudar de página e 'q' para sair do modo de navegação.)
Vamos trabalhar com algumas sequências adicionais (este levará um pouco mais de tempo para ser executado):
head -n 519 mouse.1.protein.faa > mm-second.faa
grep -c ">" mm-second.faa
Em seguida, faremos a comparação das primeiras 100 sequências:
blastp -num_threads 5 -query mm-second.faa -db zebrafish.1.protein.faa -out mm-second.x.zebrafish.txt
Você pode visualizar o arquivo de saída com:
less mm-second.x.zebrafish.txt
(E novamente, digite 'q' para sair do modo de visualização.)
Observações:
Por que levou mais tempo para fazer o BLAST em mm-second.faa do que em mm-first.faa?
Coisas para mencionar e discutir:
- Opções do blastp e -help.
- Qual valor de e-value foi usado para filtrar os resultados?
- Qual foi o valor da palavra usado para iniciar a busca?
- Opções da linha de comando, mais especificamente, por que tantas?
Automação é maravilhosa!
Por último, mas não menos importante, vamos gerar uma versão mais legível para máquinas daquele último arquivo:
blastp -num_threads 5 -query mm-second.faa -db zebrafish.1.protein.faa -out mm-second.x.zebrafish.tsv -outfmt 6
Você pode abrir o arquivo com less mm-second.x.zebrafish.tsv para ver como ele é formatado.
Em alguns casos mais do que uma proteina do zebrafish aparece no resultado. Vamos modificar os argumentos do blastp para retornar máximo uma proteina subject para cada query. Para evitar hits de fracoes das proteínas, vamos a pedir para o blastp so relatar hits onde a cobertura do query seja pelo menos 80% do seu comprimento.
blastp -num_threads 5 -query mm-second.faa -db zebrafish.1.protein.faa -subject_besthit -qcov_hsp_perc 80 -max_target_seqs 1 -out mm-second.x.zebrafish_best.tsv -outfmt '6 std qlen slen'
Por favor, revise a documentação do BLAST. Quais campos ou colunas são exibidos quando o formato de saída é configurado como outfmt 6?
blastp -help
Lembre-se de que inicialmente tínhamos 100 sequências do camundongo para realizar o BLAST. Será que todas elas resultaram em correspondências (hits) contra o peixe-zebra (zebrafish)? Como poderíamos verificar isso?
Vamos alterar o formato de saída para incluir duas colunas adicionais que mostrarão os comprimentos das sequências query e do subject. Posteriormente, utilizaremos essas informações para filtrar os resultados utilizando o comando awk, para só manter os hits onde pelo menos 80% do subject participou do alinhamento com o query.
awk '$14*0.8 <= ($10-$9+1) {print $0}' mm-second.x.zebrafish_best.tsv > mm-second.x.zebrafish_best_subject80percent.tsv
Você pode identificar quantas proteínas query se mantem nesse arquivo final? É quantas subject?