Differential Expression
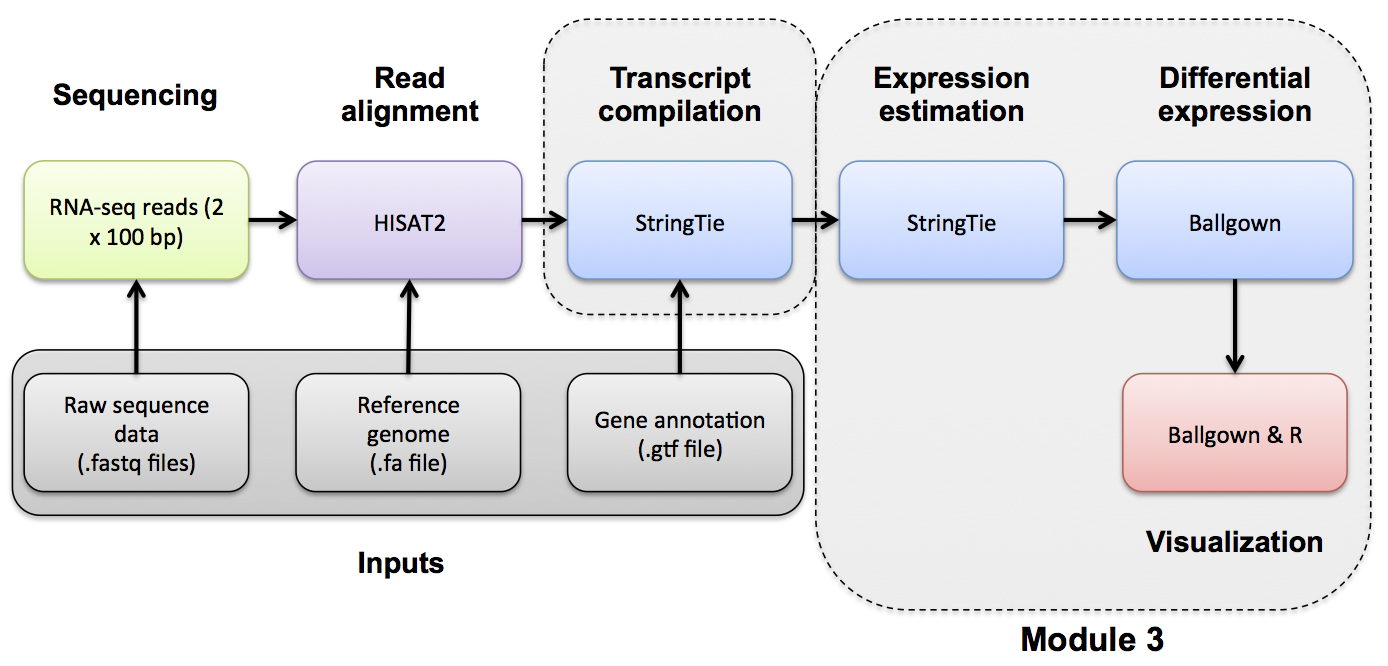
Use Ballgown to compare the tumor and normal conditions. Refer to the Ballgown manual for a more detailed explanation:
Change to ref-only directory:
mkdir -p $RNA_HOME/de/ballgown/ref_only/
cd $RNA_HOME/de/ballgown/ref_only/
Perform UHR vs. HBR comparison, using all replicates, for known (reference only mode) transcripts:
First create a file that lists our 6 expression files, then view that file, then start an R session where we will examine these results:
printf "\"ids\",\"type\",\"path\"\n\"UHR_Rep1\",\"UHR\",\"$RNA_HOME/expression/stringtie/ref_only/UHR_Rep1\"\n\"UHR_Rep2\",\"UHR\",\"$RNA_HOME/expression/stringtie/ref_only/UHR_Rep2\"\n\"UHR_Rep3\",\"UHR\",\"$RNA_HOME/expression/stringtie/ref_only/UHR_Rep3\"\n\"HBR_Rep1\",\"HBR\",\"$RNA_HOME/expression/stringtie/ref_only/HBR_Rep1\"\n\"HBR_Rep2\",\"HBR\",\"$RNA_HOME/expression/stringtie/ref_only/HBR_Rep2\"\n\"HBR_Rep3\",\"HBR\",\"$RNA_HOME/expression/stringtie/ref_only/HBR_Rep3\"\n" > UHR_vs_HBR.csv
cat UHR_vs_HBR.csv
R
A separate R tutorial file has been provided in the github repo for part 1 of the tutorial: Tutorial_Part1_ballgown.R. Run the R commands detailed in the R script.
Once you have completed the Ballgown analysis in R, exit the R session and continue with the steps below.
What does the raw output from Ballgown look like?
head UHR_vs_HBR_gene_results.tsv
How many genes are there on this chromosome?
grep -v feature UHR_vs_HBR_gene_results.tsv | wc -l
How many passed filter in UHR or HBR?
grep -v feature UHR_vs_HBR_gene_results_filtered.tsv | wc -l
How many differentially expressed genes were found on this chromosome (p-value < 0.05)?
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | wc -l
Display the top 20 DE genes. Look at some of those genes in IGV - do they make sense?
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | sort -rnk 3 | head -n 20 #Higher abundance in UHR
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | sort -nk 3 | head -n 20 #Higher abundance in HBRSave all genes with P<0.05 to a new file.
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | cut -f 6 | sed 's/\"//g' > DE_genes.txt
head DE_genes.txt
This section will compare the observed versus expected differential expression estimates for the ERCC spike-in RNAs:
cd $RNA_HOME/de/ballgown/ref_only
wget https://raw.githubusercontent.com/griffithlab/rnaseq_tutorial/master/scripts/Tutorial_ERCC_DE.R
chmod +x Tutorial_ERCC_DE.R
./Tutorial_ERCC_DE.R $RNA_HOME/expression/htseq_counts/ERCC_Controls_Analysis.txt $RNA_HOME/de/ballgown/ref_only/UHR_vs_HBR_gene_results.tsv
View the results here:
- http://YOUR_IP_ADDRESS/rnaseq/de/ballgown/ref_only/Tutorial_ERCC_DE.pdf
In this tutorial you will:
- Make use of the raw counts you generated above using htseq-count
- edgeR is a bioconductor package designed specifically for differential expression of count-based RNA-seq data
- This is an alternative to using stringtie/ballgown to find differentially expressed genes
First, create a directory for results:
cd $RNA_HOME/
mkdir -p de/htseq_counts
cd de/htseq_counts
Create a mapping file to go from ENSG IDs (which htseq-count output) to Symbols:
perl -ne 'if ($_ =~ /gene_id\s\"(ENSG\S+)\"\;/) { $id = $1; $name = undef; if ($_ =~ /gene_name\s\"(\S+)"\;/) { $name = $1; }; }; if ($id && $name) {print "$id\t$name\n";} if ($_=~/gene_id\s\"(ERCC\S+)\"/){print "$1\t$1\n";}' $RNA_REF_GTF | sort | uniq > ENSG_ID2Name.txt
head ENSG_ID2Name.txt
Determine the number of unique Ensembl Gene IDs and symbols. What does this tell you?
cut -f 1 ENSG_ID2Name.txt | sort | uniq | wc
cut -f 2 ENSG_ID2Name.txt | sort | uniq | wc
cut -f 2 ENSG_ID2Name.txt | sort | uniq -c | sort -r | head
Launch R:
R
A separate R tutorial file has been provided in the github repo for part 4 of the tutorial: Tutorial_edgeR.R. Run the R commands in this file.
Once you have run the edgeR tutorial, compare the sigDE genes to those saved earlier from cuffdiff:
cat $RNA_HOME/de/ballgown/ref_only/DE_genes.txt
cat $RNA_HOME/de/htseq_counts/DE_genes.txt
Pull out the gene IDs
cd $RNA_HOME/de/
cut -f 1 $RNA_HOME/de/ballgown/ref_only/DE_genes.txt | sort > ballgown_DE_gene_symbols.txt
cut -f 2 $RNA_HOME/de/htseq_counts/DE_genes.txt | sort > htseq_counts_edgeR_DE_gene_symbols.txt
Visualize overlap with a venn diagram. This can be done with simple web tools like:
To get the two gene lists you could use cat to print out each list in your terminal and then copy/paste.
Alternatively you could view both lists in a web browser as you have done with other files. These two files should be here:
http://YOUR_IP_ADDRESS/rnaseq/de/ballgown_DE_gene_symbols.txt http://YOUR_IP_ADDRESS/rnaseq/de/htseq_counts_edgeR_DE_gene_symbols.txt
| Previous Section | This Section | Next Section |
|---|---|---|
| Expression | Differential Expression | DE Visualization |