Alignment
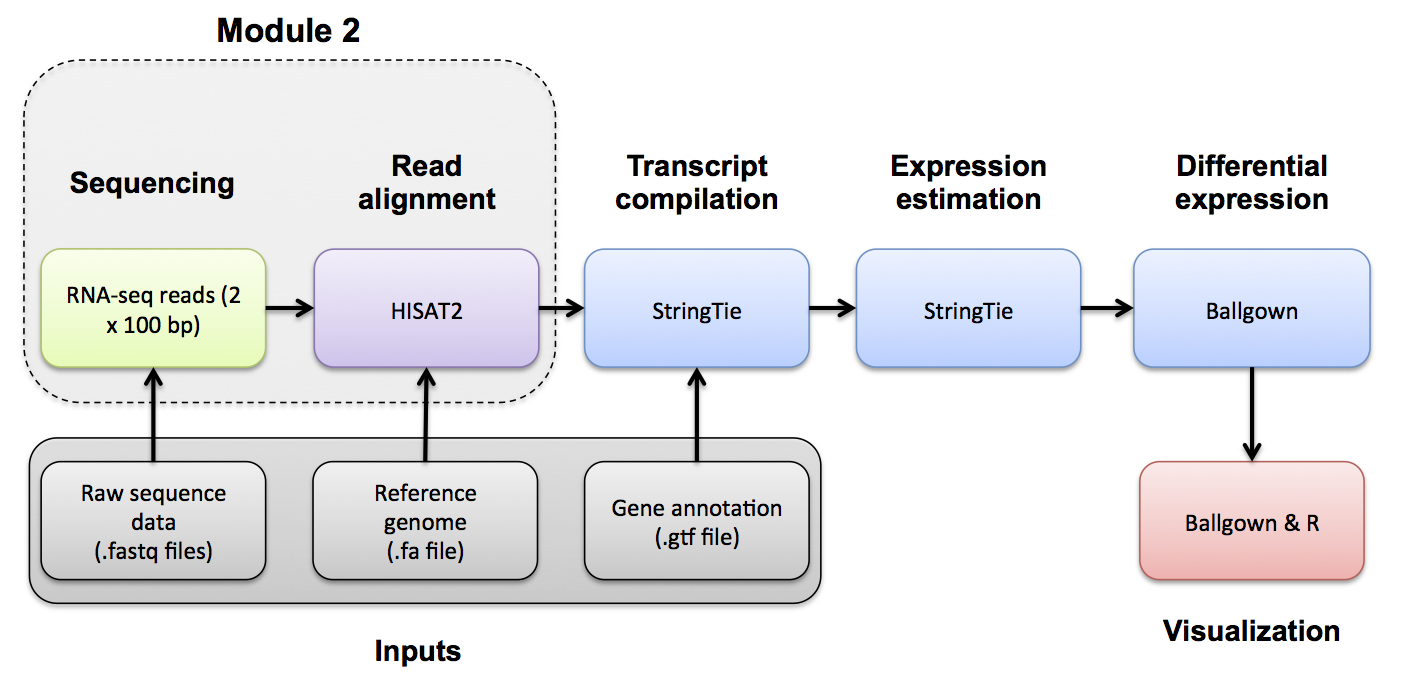
Perform alignments with HISAT2 to the genome and transcriptome.
First, begin by making the appropriate output directory for our alignment results.
echo $RNA_ALIGN_DIR
mkdir -p $RNA_ALIGN_DIR
cd $RNA_ALIGN_DIR
HISAT2 uses a graph-based alignment and has succeeded HISAT and TOPHAT2. The output of this step will be a SAM/BAM file for each data set.
Refer to HISAT2 manual for a more detailed explanation:
HISAT2 basic usage:
#hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r> | --sra-acc <SRA accession number>} [-S <sam>]
Extra options specified below:
- '-p 8' tells HISAT2 to use eight CPUs for bowtie alignments.
- '--rna-strandness RF' specifies strandness of RNAseq library. We will specify RF since the TruSeq strand-specific library was used to make these libraries. See here for options.
- '--rg-id $ID' specifies a read group ID that is a unique identifier.
- '--rg SM:$SAMPLE_NAME' specifies a read group sample name. This together with rg-id will allow you to determine which reads came from which sample in the merged bam later on.
- '--rg LB:$LIBRARY_NAME' specifies a read group library name. This together with rg-id will allow you to determine which reads came from which library in the merged bam later on.
- '--rg PL:ILLUMINA' specifies a read group sequencing platform.
- '--rg PU:$PLATFORM_UNIT' specifies a read group sequencing platform unit. Typically this consists of FLOWCELL-BARCODE.LANE
- '--dta' Reports alignments tailored for transcript assemblers.
- '-x /path/to/hisat2/index' The HISAT2 index filename prefix (minus the trailing .X.ht2) built earlier including splice sites and exons.
- '-1 /path/to/read1.fastq.gz' The read 1 FASTQ file, optionally gzip(.gz) or bzip2(.bz2) compressed.
- '-2 /path/to/read2.fastq.gz' The read 2 FASTQ file, optionally gzip(.gz) or bzip2(.bz2) compressed.
- '-S /path/to/output.sam' The output SAM format text file of alignments.
hisat2 -p 8 --rg-id=UHR_Rep1 --rg SM:UHR --rg LB:UHR_Rep1_ERCC-Mix1 --rg PL:ILLUMINA --rg PU:CXX1234-ACTGAC.1 -x $RNA_REF_INDEX --dta --rna-strandness RF -1 $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./UHR_Rep1.sam
hisat2 -p 8 --rg-id=UHR_Rep2 --rg SM:UHR --rg LB:UHR_Rep2_ERCC-Mix1 --rg PL:ILLUMINA --rg PU:CXX1234-TGACAC.1 -x $RNA_REF_INDEX --dta --rna-strandness RF -1 $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./UHR_Rep2.sam
hisat2 -p 8 --rg-id=UHR_Rep3 --rg SM:UHR --rg LB:UHR_Rep3_ERCC-Mix1 --rg PL:ILLUMINA --rg PU:CXX1234-CTGACA.1 -x $RNA_REF_INDEX --dta --rna-strandness RF -1 $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./UHR_Rep3.sam
hisat2 -p 8 --rg-id=HBR_Rep1 --rg SM:HBR --rg LB:HBR_Rep1_ERCC-Mix2 --rg PL:ILLUMINA --rg PU:CXX1234-TGACAC.1 -x $RNA_REF_INDEX --dta --rna-strandness RF -1 $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./HBR_Rep1.sam
hisat2 -p 8 --rg-id=HBR_Rep2 --rg SM:HBR --rg LB:HBR_Rep2_ERCC-Mix2 --rg PL:ILLUMINA --rg PU:CXX1234-GACACT.1 -x $RNA_REF_INDEX --dta --rna-strandness RF -1 $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./HBR_Rep2.sam
hisat2 -p 8 --rg-id=HBR_Rep3 --rg SM:HBR --rg LB:HBR_Rep3_ERCC-Mix2 --rg PL:ILLUMINA --rg PU:CXX1234-ACACTG.1 -x $RNA_REF_INDEX --dta --rna-strandness RF -1 $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz -2 $RNA_DATA_DIR/HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz -S ./HBR_Rep3.sam
Note: in the above alignments, we are treating each library as an independent data set. If you had multiple lanes of data for a single library, you could align them all together in one HISAT2 command. Similarly you might combine technical replicates into a single alignment run (perhaps after examining them and removing outliers...). To combine multiple lanes, you would provide all the read1 files as a comma separated list for the '-1' input argument, and then all read2 files as a comma separated list for the '-2' input argument, (where both lists have the same order) : You can also use samtools merge to combine bam files after alignment. This is the approach we will take.
HISAT2 generates a summary of the alignments printed to the terminal. Notice the number of total reads, reads aligned and various metrics regarding how the reads aligned to the reference.
Convert HISAT2 sam files to bam files and sort by aligned position
samtools sort -@ 8 -o UHR_Rep1.bam UHR_Rep1.sam
samtools sort -@ 8 -o UHR_Rep2.bam UHR_Rep2.sam
samtools sort -@ 8 -o UHR_Rep3.bam UHR_Rep3.sam
samtools sort -@ 8 -o HBR_Rep1.bam HBR_Rep1.sam
samtools sort -@ 8 -o HBR_Rep2.bam HBR_Rep2.sam
samtools sort -@ 8 -o HBR_Rep3.bam HBR_Rep3.sam
Make a single BAM file combining all UHR data and another for all HBR data. Note: This could be done in several ways such as 'samtools merge', 'bamtools merge', or using picard-tools (see below). We chose the third method because it did the best job at merging the bam header information. NOTE: sambamba also retains header info.
cd $RNA_HOME/alignments/hisat2
java -Xmx2g -jar $RNA_HOME/tools/picard.jar MergeSamFiles OUTPUT=UHR.bam INPUT=UHR_Rep1.bam INPUT=UHR_Rep2.bam INPUT=UHR_Rep3.bam
java -Xmx2g -jar $RNA_HOME/tools/picard.jar MergeSamFiles OUTPUT=HBR.bam INPUT=HBR_Rep1.bam INPUT=HBR_Rep2.bam INPUT=HBR_Rep3.bam
Count the alignment (BAM) files to make sure all were created successfully (you should have 8 total)
ls -l *.bam | wc -l
ls -l *.bam
Assignment: Perform some alignments on additional read data sets. Align the reads using the skills you learned above. Try using the HISAT2 aligner. Also practice converting SAM to BAM files, and merging BAM files.
- Hint: Do this analysis on the additional data and in the separate working directory called ‘practice’ that you created in Practical Exercise 3.
Questions
- What is the difference between a .sam and .bam file?
- If you sorted the resulting BAM file as we did above, is the result sorted by read name? Or position?
- Which columns of the BAM file can be viewed to determine the style of sorting?
- What command can you use to view only the BAM header?
Solution: When you are ready you can check your approach against the Solutions
| Previous Section | This Section | Next Section |
|---|---|---|
| Adapter Trim | Alignment | IGV |