MultiQC v1.25.2 - 2024-11-20
Multiple bug fixes and minor updates.
- Add natural sort for sample sorting (#2959)
- Custom content: for
plot_type: image, supportcustom_dataconfig with section name and description. Fix misleading logging (#2939) - Config validation improvements (group messages, cast types, validate column headers) (#2899)
- Workaround for displaying sample grouping in Safari because of missing
visibility: collapse(#2941) - Fix table CSV export where a title contains a comma (#2911)
- Showing table in notebooks: respect
col1_header(#2914) - Customizing
custom_table_header_config: fix docs, support both the old and the new ways (#2955) - Table scatter mini-plots: fix rounding and range (#2956)
- File line block iterator: fix reading long lines that do not fit one block (#2935)
- Fix
cond_formatting_rulestype hint to avoid validation error (#2922) - Fix
config.prepend_dirsor-d -dd 1(#2913) - Sample grouping fixes (#2920):
- Keep sample name column fix width to avoid jumping
- Fix hiding columns through the modal
- Custom content fixes:
- ngsbits: add submodule samplegender (#2854)
- nanoq: change lineplots for barplots (#2934)
- Qualimap: clarify the direction of the transcript in coverage plot (#2946)
- picard: add table with all metrics to VariantCallingMetrics section (#2885)
- Nanostat: add general stats columns (#2961)
- Samtools: add insert size to general stats table (#2905)
- bcl2fastq: fix missing
R1_*/R2_*metrics (#2965) - Cutadapt: fix for null values from r2 data (#2936)
- Qualimap: fix parsing ∞ value (#2937)
- bclconvert: fix undetermined barcodes plot (#2976)
- featurecounts: fix missing section name and anchor (#2967)
- Pin kaleido to 0.2.1 (new 0.4.1 does not embed a browser and thus not portable) (#2963)
MultiQC v1.25.1 - 2024-09-30
Python 3.13, bugs fixed, improved sample grouping UI, and handling freezes in containers with incompatible architectures.
- Support Python 3.13 (officially to be released on Oct 7). Python 3.8 is supported for now, but might drop support in future releases, so make sure you update! (#2871)
- Table sample groups UI: allow clicking the entire row to expand, add cursor pointer (#2871)
- Disable plot export in incompatible architecture containers (when running through rosetta) (#2888)
- Fix export general stats to
multiqc_data.json: flatten row groups for back-compatibility (#2879) - Custom content:
- Kraken:
- bbmap: support qhist outputs with only R1 and extra header (#2882)
- Picard HsMetrics: fix collecting data sources (#2880)
- Test Docker image builds on every PR commit (#2886)
- Suppress "SyntaxWarning: invalid escape sequence" warnings from
colormath(#2889) - Check the
add_data_sourceargs (eitherpathorfshould be specified), usestrict_helpers.lint_error(#2865)
MultiQC v1.25 - 2024-09-16
New feature: grouping samples in the General Statistics table.

Some modules - prominently FastQC - may produce multiple results per sample, e.g. for the forward and the reverse reads. To group such results in the table together, a new configuration option is introduced.
This feature is currently opt-in, you'll need to set table_sample_merge in a MultiQC config file to use (see docs above). We'd love to hear your feedback! We hope to enable it by default for common file suffix patterns in a future release.
Because MultiQC needs to know how to merge each column (sum, average, etc), each module must implement it independently. Currently it's supported by the FastQC and Cutadapt modules. If you'd like support added to another module, please let us know in a GitHub issue. Details of how to add it into module code can be found in the moduel development documentation.
The new box plot type, added in v1.21, are now available to use with custom content! See 2847 for configuration examples.
- Group read pairs in general stats (#2794, #2848)
- Support boxplot in custom content (#2847)
- Allow
x_band,x_lines,x_minrangefor any plot type (specifically, scatter plots) (#2851) - When both
contentsandcontents_reare specified in a search patterns, treat it as logical AND (#2828)
- NanoStat: support multi-sample logs (#2852)
- Samtools coverage: support
exclude_contigsandinclude_contigs(#2840) - Ganon: support non-verbose output, fix missing
removed with --min-count(#2838) - fastp: support the
--mergedflag (#2834)
- Workaround for the hanging Kaleido duing plot export: run plot export in separate threads with a timeout and fallback on freezes, add try-catch for crashes (#2836, #2819)
- Fix copying
multiqc.loginto themultiqc_dataoutput folder (#2829) - Fix applying
exclude_filesin search patterns (#2804) - Fix bar plot export from toolbox (#2845)
- Fix autoselection in plot export toolbox (#2844)
- Fix
config.replace_samplesfor custom content genstats table (#2841)
- Picard: fix parsing Sentieon insert size metrics (#2823)
- Picard: fix a search pattern of CollectRnaSeqMetrics (#2811)
- Kraken: fix for empty top ranks. Also handle it in bargraph (#2822)
- Cellranger: fix data source tag (#2821)
- FastQC: fix the
Per Base Sequence Contentdetail plot click when a module was run multiple times (#2856) - FastQC: fix calculating average read length (#2817)
- Separate anchors and IDs for sections: use IDs for Python configation, and anchors in HTML (#2797, #2833)
- Prefix table column IDs with namespace, but allow configuration to use both short and long anchors (#2818)
- More type hinting in plots (#2816, #2850)
- Use
typing.NewTypefor all Python versions (#2820) - Remove
pyaml_envdependency and apply the fix for theSyntaxWarning(#2837)
- Add
py.typedto mark package as providing type information (#2846)
MultiQC v1.24.1 - 2024-08-21
A bug fix release mainly to restore compatibility with Python 3.8. Aside from that, few other minor bug fixes:
- FastQC: fix long-standing issue misplacing status labels when
anchoris specified in the custom config (#2790) - Freyja: handle empty inputs, and ensure deterministic sample order (#2788)
- Allow numeric xcats and ycats for the heatmap plot (#2787)
- Make sure that config's
extra_fn_clean_extsandfn_clean_extsdon't conflict when both specified (#2783)
MultiQC v1.24 - 2024-08-19
Mostly a maintenance release, containing several bugfixes, performance improvements, plus 6 new modules, along with improvements of the existing modules.
The most significant performance boost got the Kraken and Mosdepth modules, that now don't take way more memory and CPU than any other typical module:
| Tool | Data Set | Memory - Before | CPU - Before | Memory - After | CPU - After |
|---|---|---|---|---|---|
| mosdepth | 1 set of files | 196 Mb | 3.04s | 129 Mb | 2.27s |
| 10 | 464 Mb | 8.48s | 131 Mb | 6.11s | |
| 100 | 3,719 Mb | 63.19s | 172 Mb | 43.12s | |
| kraken | 1 set of files | 155 Mb | 2.07s | 132 Mb | 2.20s |
| 10 | 606 Mb | 8.39s | 180 Mb | 3.47s | |
| 100 | 4,970 Mb | 71.89s | 809 Mb | 14.53s |
Large plots that may hang browser are now not loaded by default, and the user can click
a button to load, so the heavy plots don't slow down the initial report rendering. This
is controlled by the config.plots_defer_loading_numseries: 100 option.
- Search patterns: allow multiple values for
contents(#2696) - Custom content:
- Plots:
- Defer render of plots if number of samples >
config.plots_defer_loading_numseries(#2759, #2777, #2773, #2774) - Line plot: show markers when num of data points <
config.lineplot_number_of_points_to_hide_markers(=50) (#2760). As a nice consequence, trivial lines of a single data point become visible. - Line plot: smooth by default to 500 points on the X axis to avoid inflating the report file size (#2776)
- Allow to configure the scale of the exported plot fonts through the config option
config.plots_export_font_scale(#2758) - Improve the performance of loading large tables in browser (#2737)
- Fix the toolbox highlight of the line plots (#2724)
- Defer render of plots if number of samples >
- The function that returns built plots in an interactive session now uses the module anchor (or lowercase module name) to key the results (#2741)
- More helpful config validation error: print the parent model name, if applicable (#2709)
- VG (#2690), a toolkit to manipulate graphical genomes. The module parses vg-stats reports that summarize alignment stats from a GAM file.
- ngs-bits (#2231). A tool that calculating statistics from FASTQ, BAM, and VCF files. The module parses XML output generated for two tools in the ngs-bits collection:
- Pairtools (#1148). A toolkit for Chromatin Conformation Capture experiments. Handles short-reads paired reference alignments, extracts 3C-specific information, and perform common tasks such as sorting, filtering, and deduplication. The module parses summary statistics generated by pairtools's
dedupandstatstools. - nanoq (#2723). A tool that reports read quality and length from nanopore sequencing data.
- Ganon (#1935). A tool for metagenomics classification: quickly assigns sequence fragments to their closest reference among thousands of references via Interleaved Bloom Filters of k-mer/minimizers
- Fix
--pdfoption to generatemultiqc_report.pdf(#2733) - Fix saving table plots to file (#2735)
- Fix adding software versions when
config.run_modulesis set (#2755) - Fix toolbox highlight in line plots (#2724)
- Refactor
write_resultsto avoid dynamically overridingconfig, fixesmodule.write_data_file(#2722) - Search stats: do not double-count ignored files (#2708)
- Escape values passed to HTML properties (e.g.
valin tables) (#2706) - Fix re-loading explicit user configs in interactive sessions (#2704)
- Fix file search performance regression (#2762)
- Fix handling module href string (#2739)
- Custom content:
- Kraken: optimize memory and runtime (#2756)
- Mosdepth: optimize memory and runtime (#2748, #2749)
- Abstract
config.get_cov_thresholdsfunction formosdepthandqualimap(#2707) - Anglerfish: adjust for version 0.6.1 (#2757)
- Umitools: prefer output for sample name, handle the
<stdin>/<stdout>placeholders (#2698) - Kraken: fix top % calculation, more efficient total read count calculation (#2744)
- Bracken: when printing number of found samples, indicate that running Bracken not Kraken (#2743)
- Nonpareil: Update docs about new version that generates the JSON file (#2734)
- STAR: add all alignment summary metrics into a new separate table (#1828)
- Pairtools: fix typos and grammar, remove redundancies (#2711)
- Peddy sex plot: color predicted sex (#2778)
- FastQC: for plots with bp on the X axis, use the interval start instead of average (#2790)
- Cellranger: fix for missing
analysis_tabdata (#2771) - Fix setting coverage thresholds for
mosdepthandqualimap(#2754) - Nonpareil: fix running with >12 samples (#2752)
- Bracken: fix bug when direct reads not classified (#2738)
- ngsderive: fix ValueError in the
encodingsubmodule (#2740) - RSeQC: fix duplicated namespace (#2732)
- Glimpse: fix parsing data, add proper type hints (#2721)
- Fix ignoring samples in
spaceranger,ngsbits,isoseq,dragen coverage(#2717) - Glimpse: clean and fix filtering samples (#2716)
- Spaceranger: fix ignoring samples (#2714)
- Refactor
write_resultsto avoid dynamically overridingconfig, fixesmodule.write_data_file(#2722) - Modules:
- isoseq and odgi: fix module warnings and error handling (#2718)
- Qualimap: refactor and add type hints (#2707)
- FastQC: refactor and add type hints (#2763)
- Kraken: refactor and add type hints (#2744)
- Cell Ranger: refactor, add type hints, get rid of module mixins and fields (#2775)
- Spaceranger: refactor, add type hints, get rid of module mixins and fields (#2714)
- RSeQC: refactor, add type hints, and remove
multiqc_rseqc.js(#2710)
- Split up the tests for sample versions discovery (#2751)
- Move integration test variations into unit tests, actually test them (#2713)
- Move module docs to the docstrings & generate
docs/modules/*.mdfrom the docstrings using a separate script (#2703) - Embed the search patterns into the module docs (#2765)
- Dockerfile: use
COPYinstead ofADDto copy only relevant files, update base image to Python 3.12 (#2700)
MultiQC v1.23 - 2024-07-09
Bug fixes, integration of pytest and mypy, and one new module.
From the user perspective, this is mostly a maintenance release, containing several important bugfixes, plus minor improvements and a new module - Glimpse.
For developers, there are two significant additions to the CI workflow:
- pytest, along with unit tests covering the core library,
- and mypy, along with ensuring that the core codebase is fully type-annotated.
The core unit tests are located in the multiqc/tests folder, and the module tests are
located in the corresponding multiqc/modules/*/tests subfolders.
The CI workflows
are refactored to separate the integration tests and the unit tests, to improve the
granularity and parallelization. The tests are discovered and executed with pytest,
and the coverage is reported by codecov.
The multiqc/tests subfolder has several test files that cover most of the core library.
It also has a test_modules_run.py
tests that checks that every module didn't crash when being run on the corresponding data
in test-data, and added something into the report.
That is somewhat of a blanket test for modules, that doesn't check if the modules logic
worked correctly. For that reason, the users are encouraged to write more comprehensive
tests that take the specific module logic into account, and place them in
multiqc/modules/*/tests. For some initial examples, consider checking:
- The samtools flagstat
test that verifies some logic in the
flagstatsubmodule of thesamtoolsmodule; - The picard tools test that checks that every submodule for each Picard tool worked correctly.
- Custom content"
- Re-enabling the
software_versionmodule section (#2670) - When
--no-ansiis set, disable colors inrich_clicktoo (#2678) - Support CWD path filters (
./path/...) in config (#2676) - Fix writing report to stdout with
--filename stdout, log to stderr (#2672) - Interactive use:
- Run mypy on core library (#2665)
- Add tests for plot export (#2682)
- Add tests for command line use, including for passing
TMPDIR(#2677) - Custom content: allow hash-fenced table columns (#2649)
- Software versions: parse for sorting, but preserve the original strings (#2671)
- Allow both table-level and column-level custom plot config for table (#2662)
- Glimpse (#2492)
- Fix parsing kraken vs. bracken: respect
num_linesin search patterns (#2657) - Fix the
bbmap/qchistsearch pattern (#2661)
- Picard HsMetrics: support any custom X coverage metrics (#2663)
- Samtools coverage: avoid hard crash for invalid file contents (#2664)
- Abstract code related to temporary directory creation into a separate module (#2675)
- Use pull-request labels and milestones for changelog generation (#2691)
MultiQC v1.22.3 - 2024-06-22
Contains fixes of multiple bugs collected after the last release, along with few minor improvements.
- Fix the
re_contentssearch patterns when pattern is found in the middle of the file. Fixes finding logs from several Picard submodules, likeCollectRnaSeqMetricsandCollectWgsMetricsin some cases (#2610) - Fixes the
run_modulesoption use when the module anchor doesn't match the module entry point ID (e.g.DRAGENanddragen) (#2633) - Fix use of custom search patterns for custom content (#2647)
- Fix plot export with
export_plots: trueor--export(#2637) - Correctly handle old-style
labelsections inx_linesory_linesin line plot configs (#2648) - Fix disabling
sort_rowsin custom content by subclassingTableConfigfromValidatedConfigand use deprecated (#2604) - When user provides a search pattern dictionary in config, recursively update instead of replacing (#2620)
- Fix config update when dict replaced with list, e.g. a
search_patternsitem is a list that's replaced with a dict (https://github.com/MultiQC/MultiQC/commit/c388178bb6d9f143c6f8e8b0146647a067021ea4)
- Add unit tests for core and some modules (see
picardorsamtools), as well ascodecovreport (#2624)- Now MultiQC checks if every module does something productive with the provided test data in
test-data. - For modules with many submodules (picard, dragen), additionally check if every submodule parses the expected number of samples from
test-datafiles. - Users can put module tests in
testsubfolders, e.g. https://github.com/MultiQC/MultiQC/tree/main/multiqc/modules/picard/tests - Use
pytestfor all core unit tests (#2623) - Move unit tests from the
test-datarepo intotestsfolder (#2622)
- Now MultiQC checks if every module does something productive with the provided test data in
- Plot config validation:
- Validate line plot
x_lines,x_bands, etc. with a Pydantic model, includinglabelsubsections (#2648) - Validate line plot series and
extra_serieswith a Pydantic model (#2573) - Validate table config (#2604)
- Make the "unrecognised field" error a warning
- Rename deprecated plot config fields in internal modules (#2636)
- Validate line plot
- Show progress bar for exporting flat plot images (#2639)
- Better error message for incorrect
run_modules(#2635) - Increase flat plots sample number threshold to 1000 (#2615)
- Small speed-up of the line block iterator (#2588)
- Update README logos for better compatibility (#2603)
- Docs: don't use raw markdown links (#2642)
- Allow to override
showlegendfor line config plots. Default to not-show for large datasets to avoid bloated legends (#2615) - Show error message if failed to parse custom content header (https://github.com/MultiQC/MultiQC/commit/d736846a0c23410243f80b2bdca984363211ffc3)
- Load every found config file once https://github.com/MultiQC/MultiQC/commit/422b39bc787720cefea81a85dabbf6411b3421ac
- Picard
- Fix finding
CollectRnaSeqMetricsandCollectWgsMetricslogs by fixing there_contentssearch patterns (#2610)
- Fix finding
- biobambam2
- Fix parsing
markdupslogs
- Fix parsing
- DRAGEN
- FastQC
- Default to
showlegend: false, as we don't distinguish the sample colors, unlessfastqc_config: status_checks: false'is set (#2615)
- Default to
- BBTools
- Fix incorrect calculation of % Q30 Bases (#2628)
- Samtools
markdup: resolve inconsistent non-optical pair duplicate variable name in samtools markdup module (#2626)
- NanoStat
- Support different
Qcutoffs (#2645)
- Support different
- Salmon
- Fix ignored parsed
library_typeswhen its type is list (#2617)
- Fix ignored parsed
- UMI-tools
- Improve
extractplots (#2614)
- Improve
- BCL Convert
- Fix 'pecent' typo (#2612)
MultiQC v1.22.2 - 2024-05-31
Bug fix release. Two major issues are fixed here:
- Fixed running same module twice with
path_filters(e.g. trimmed vs. raw FastQC), - The raw data
report_saved_raw_datais re-added in multiqc_data.json by default.
- Fix running same module multiple times in the report (e.g. trimmed vs. raw FastQC) (#2592)
- Preserve
report_saved_raw_datain multiqc_data.json by keepingpreserve_module_raw_data: falseby default (#2591) - Table headers: do not set namespace to
Nonewhen there is single namespace (#2590) - Re-enable falling back to flat plots for large datasets (#2580)
- Reset in
multiqc.run(*)to allow running it twice interactively (#2598) - Fix scatter plot in
--flatmode when there are categorical axes (#2600) - Fix hiding table column with all empty values in custom content (#2599)
- Table "Copy" button: include headers (#2594)
- QUAST
- Underscore attributes captured by lambdas to avoid wiping them after module finished (#2581)
- Cell Ranger
- Handle missing
vdj_annotationandvdj_enrichmentsections (#2579)
- Handle missing
- fgbio
- Fix links in fgbio.md (#2586)
- Glimpse:
- Add support for Glimpse concordance metrics #2491
- Custom content
- Support DOI for custom content (#2582)
MultiQC v1.22.1 - 2024-05-17
This bug fix release addresses the file search problem when MultiQC executed as a typical Nextflow job. See #2575 for detail.
MultiQC v1.22 - 2024-05-14
Version 1.22 brings some major behind-the-scenes refactoring to MultiQC. This unlocks a number of new features, such as the ability to use MultiQC as a Python library in scripts / notebooks, and run-time validation of plot config attributes.
This release also introduces some huge performance improvements thanks to @rhpvorderman. Compared to v1.21, a typical v1.22 run is 53% faster and has a 6x smaller peak-memory footprint - well worth updating!
Finally, support for the depreciated HighCharts plotting library is fully removed in v1.22, bringing to a close a long standing project to migrate to Plotly.
For more information, please see the MultiQC release blog article on the Seqera website
- Remove the
highchartstemplate and Highcharts and Matplotlib dependencies (#2409) - Remove CSP.txt and the linting check, move the script that prints missing hashes under
scripts. Admins of servers with Content Security Policy can use it to print missing hashes when they install a new MultiQC version with:python scripts/print_missing_csp.py --report full_report.html(#2421) - Do not maintain change log between releases (#2427)
- Use native clipboard API (#2419)
- Profile runtime: visualize per-module memory and run time (#2548, #2547)
- Refactoring for performance:
- Search file blocks rather than individual lines for faster results (#2513)
- Refactor file content search for a 40% speed increase (#2505)
- Sort
filepatternsfor faster searching (#2506) - Use
array.arrayfor in-memory plot data, stream to render Jinja and dump JSON to reduce memory requirement (#2515) - Speed up all modules by caching
spectra.scaleand using sets instead of lists (#2509) - Stream json data to a file to save 30% of the memory (#2510)
- Do
replace_nanin place rather than creating a new object (#2529) - Use gzip rather than lzstring for compression and decompression of the plot data (#2504)
- Use gzip level 6 for faster json compression (#2553)
- Clean up module raw data after running each module, significantly reduces the memory footprint (#2551)
- Refactoring for interactivity and validation:
- Top-level functions for MultiQC use as a library (#2442)
- Pydantic models for plots and datasets (#2442)
- Validating plot configs with Pydantic (#2534)
- Use dataclasses for table and violin columns (#2546)
- Break up the main run function into submodules (#2446)
- Deprecate
multiqc.utils.configandmultiqc.utils.reportin favour ofmultiqc.configandmultiqc.report(#2542) - Static typing of the report and config modules (#2445)
- Add type hints into core codebase (#2434)
- Consistent config options: rename
decimalPlacestott_decimals(#2451) - Remove encoding and shebang headers from module files (#2425)
- Refactor line plot categories: keep boolean throughout the code, and data points as pairs for simplicity (#2418)
- Fixes:
- Fix error when using default sort (#2544)
- Do not attempt to render flat plot when no data (#2490)
- Fix export plots with
--exportand always export data (#2489) - Fix: make sure
modifylambda not present in JSON dump (#2455) - Enable
--exporteven when writing interactive plots (#2444) - Replace
NaNwithnullin exported JSON (#2432) - Fix
y_minrangeoption (#2415)
- Reduce report size: exclude plot data for sections in
remove_sections(#2460) - Add
geandletocond_formatting_rules(#2494) - CI: use
uv pip(#2352) - Lint check for use of
f["content_lines"](#2485) - Allow to set style of line graph (
linesorlines+markers) per plot (#2413) - Add
CMDtoDockerfileso a default run without any parameters displays the--help(#2279) - Custom content tables are sorted by key by default (unless
sort_rows: falseis set in config), to harmonize with tables in modules.
- Hostile (#2501)
- New module: Hostile is a short and long host reads removal tool
- Sequali (#2441)
- New module: Sequali Universal sequencing QC
- Adapter Removal
- Standardize module names: use the came case (#2433)
- Bamdst
- BBTools
- Set missing values to
Noneforbbmap qahist(#2411)
- Set missing values to
- Bcftools
- Stats: add multialleic sites column (#2414)
- BCL Convert
- Busco
- Fix barplot colors (#2453)
- Cell Ranger
- Fix parsing antibody tab without
antibody_treemap_plot(#2525)
- Fix parsing antibody tab without
- Cutadapt
- Speed up module by caching parsing versions (#2528)
- DRAGEN
- Add ploidy estimation table (#2496)
- fastp
- When could not parse sample name from command (i.e.
stdin), use filename and proceed (#2536)
- When could not parse sample name from command (i.e.
- FastQC
- Skip per tile sequence quality section in FastQC reports for better performance (#2552)
- Fix a
ZeroDivisionErrorerror (#2462) - Fix memory leak to make 7 times faster and use 10 times less memory (#2552)
- Do not keep intermediate data in memory to reduce memory footprint further (#2516 )
- Add option to ignore FastQC quality thresholds (#2486)
- goleft indexcov
- Work correctly even if no valid contigs in input (#2540)
- mosdepth
- Fix absolute coverage plot (#2488)
- nonpareil
- Change write_data_file label to be consistent with other modules (#2472)
- Picard
- qc3C
- Fix detecting sample name for relative path (#2502)
- QualiMap
- BamQC: when trimming long tails, keep at least 20x (#2431)
- Samtools
- Space Ranger
- fix for missing
genomic_dnasection (#2429)
- fix for missing
- xengsort
- Fix parsing long files (do no use
content_lines) (#2484)
- Fix parsing long files (do no use
MultiQC v1.21 - 2024-02-28
Added a new plot type: box plot. It's useful to visualise a distribution when you have a set of values for each sample.
from multiqc.plots import box
self.add_section(
...,
plot=box.plot(
{
"sample 1": [4506, 4326, 3137, 1563, 1730, 3254, 2259, 3670, 2719, ...],
"sample 2": [2145, 2011, 3368, 2132, 1673, 1993, 6635, 1635, 4984, ...],
"sample 3": [1560, 1845, 3247, 1701, 2829, 2775, 3179, 1724, 1828, ...],
},
pconfig={
"title": "Iso-Seq: Insert Length",
},
)
)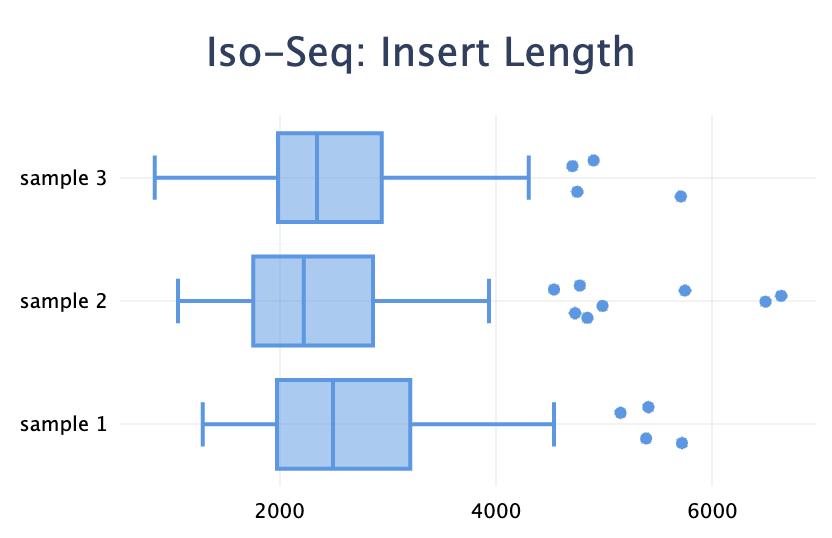
Note the difference with the violin plot: the box plot visualises the distributions of many values within one sample, whereas the violin plot shows the distribution of one metric across many samples.
The setup.py file has been superseded by pyproject.toml for the build configuration.
Note that now for new modules, an entry point should be added to pyproject.toml instead of setup.py, e.g.:
[project.entry-points."multiqc.modules.v1"]
afterqc = "multiqc.modules.afterqc:MultiqcModule"The heatmap plot now supports passing a dict as input data, and also supports a zlab
parameter to set the label for the z-axis:
from multiqc.plots import heatmap
self.add_section(
...,
plot=heatmap.plot(
{
"sample 1": {"sample 2": 0, "sample 3": 1},
"sample 2": {"sample 1": 0, "sample 3": 0},
"sample 3": {"sample 1": 1, "sample 2": 0, "sample 3": 1},
},
pconfig={
"title": "Sample comparison",
"zlab": "Match",
},
)
)- New plot type: box plot (#2358)
- Add "Export to CSV" button for tables (#2394)
- Replace
setup.pywithpyproject.toml(#2353) - Heatmap: allow a dict dicts of data (#2386)
- Heatmap: add
zlabconfig parameter. Showxlab,ylab,zlabin tooltip (#2387) - Warn if
run_modulescontains a non-existent module (#2322) - Catch non-hashable values (dicts, lists) passed as a table cell value (#2348)
- Always create JSON even when MegaQC upload is disabled (#2330)
- Use generic font family for Plotly (#2368)
- Use a padded span with
nowrapinstead of before suffixes in table cells (#2395) - Refactor: fix unescaped regex strings (#2384)
Fixes:
- Pin the required Plotly version and add a runtime version check (#2325)
- Bar plot: preserve the sample order (#2339)
- Bar plot: fix inner gap in group mode (#2321)
- Violin: filter
Infvalues (#2380) - Table: Fix use of the
no_violin(ex-no_beeswarm) table config flag (#2376) - Heatmap: prevent from parsing numerical sample names (#2349)
- Work around call of
full_figure_for_developmentto avoid Kaleido errors (#2359) - Auto-generate plot
idwhenpconfig=None(#2337) - Fix: infinite
dmaxordminfail JSON dump load in JavaScript (#2354) - Fix: dump
pconfigfor MegaQC (#2344)
- IsoSeq
- Iso-Seq contains the newest tools to identify transcripts in PacBio single-molecule sequencing data (HiFi reads).
clusterandrefinecommands are supported.
- Iso-Seq contains the newest tools to identify transcripts in PacBio single-molecule sequencing data (HiFi reads).
- Space Ranger
- Works with data from 10X Genomics Visium. Processes sequencing reads and images created using the 10x Visium platform to generate count matrices with spatial information.
- New MultiQC module parses Space Ranger quality reports.
- bcl2fastq: fix the top undetermined barcodes plot (#2340)
- DRAGEN: add few coverage metrics in general stats (#2341)
- DRAGEN: fix showing the number of found samples (#2347)
- DRAGEN: support
gvcf_metrics(#2327) - fastp: fix detection of JSON files (#2334)
- HTSeq Count: robust file reading loop, ignore
.parquetfiles (#2364) - Illumina InterOp Statistics: do not set
'scale': Falseas a default (#2350) - mosdepth: fix regression in showing general stats (#2346)
- Picard: Crosscheck Fingerprints updates (#2388)
- add a heatmap for LOD scores besides a table
- if too many pairs in table, skip those with
Expectedstatus - use the
warnstatus forInconclusive - add a separate sample-wise table instead of general stats
- sort tables by status, not by sample name
- add a column "Best match" and "Best match LOD" in tables
- hide the LOD Threshold column
- PURPLE: support v4.0.1 output without
versioncolumn (#2366) - Samtools: support new
coveragecommand (#2356) - UMI-tools: support new
extractcommand (#2296) - Whatshap: make robust when a stdout is appended to TSV (#2361)
MultiQC v1.20 - 2024-02-12
MultiQC v1.20 comes with totally new plotting code for MultiQC reports. This is a huge change to the report output. We've done our best to maintain feature parity with the previous plotting code, but please do let us know if you spot any bugs or changes in behaviour by creating a GitHub issue.
This change comes with many improvements and new features, and paves the way for more in the future. To find out more, read the associated blog post.
For now, you can revert to the previous plotting code by using the highcharts report template (multiqc --template highcharts). This will be removed in v1.22.
Note that there are several plotting configuration options which have been removed:
click_funccursortt_percentages(usett_suffix: "%")- Bar plot:
use_legend(automatically hidden if there is only 1 category)
- Line plot:
labelSizexDecimals,yDecimals(automatic if all values can be cast to int)xLabelFormat,yLabelFormat(usett_label)pointFormat
- Heatmap:
datalabel_colourborderWidth
The v1.20 release is also the first release we've had since we moved the MultiQC repositories. Please note that the code is now at MultiQC/MultiQC (formerly ewels/MultiQC) and the same for the Docker repository. The GitHub repo should automatically redirect, but it's still good to update any references you may have.
- Support Plotly as a new backend for plots (#2079)
- The default template now uses Plotly for all plots
- Added a new plot type
violin(replacesbeeswarm) - Moved legacy Highcharts/Matplotlib code under an optional template
highcharts(#2292)
- Move GitHub repository to
MultiQCorganisation (#2243) - Update all GitHub actions to their latest versions (#2242)
- Update docs to work with Astro 4 (#2256)
- Remove unused dependency on
futurelibrary (#2258) - Fix incorrect scale IDs caught by linting (#2272)
- Docs: fix missing
vprefix in docker image tags (#2273) - Unicode file reading errors: attempt to skip non-unicode characters (#2275)
- Heatmap: check if value is numeric when calculating min and max (#2276)
- Add
filesearch_file_sharedconfig option, remove unnecessary per-modulesharedflags in search patterns (#2227) - Use alternative method to walk directory using pathlib (#2277)
- Export
config.output_dirin MegaQC JSON (#2287) - Drop support for module tags (#2278)
- Pin
Pillowpackage, wrap add_logo in try-except (#2312) - Custom content: support multiple datasets (#2291)
- Configuration: fix reading config.output_fn_name and --filename (#2314)
- Bamdst (#2161)
- Bamdst is a lightweight tool to stat the depth coverage of target regions of bam file(s).
- MetaPhlAn (#2262)
- MetaPhlAn is a computational tool for profiling the composition of microbial communities from metagenomic shotgun sequencing data.
- MEGAHIT (#2222)
- MEGAHIT is an ultra-fast and memory-efficient NGS assembler
- Nonpareil (#2215)
- Estimate metagenomic coverage and sequence diversity.
- Bcftools: order variant depths plot categories (#2289)
- Bcftools: add missing
self.ignore_samplesin stats (#2288) - BCL Convert: add index, project names to sample statistics and calculate mean quality for lane statistics. (#2261)
- BCL Convert: fix duplicated
yieldfor 3.9.3+ when the yield is provided explicitly in Quality_Metrics (#2253) - BCL Convert: handle samples with zero yield (#2297)
- Bismark: fix old link in docs (#2252)
- Cutadapt: support JSON format (#2281)
- HiFiasm: account for lines with no asterisk (#2268)
- HUMID: add cluster statistics (#2265)
- mosdepth: add additional summaries to general stats #2257 (#2257)
- Picard: fix using multiple times in report: do not pass
module.anchortoself.find_log_files(#2255) - QualiMap: address NBSP as thousands separators (#2282)
- Seqera Platform CLI: updates for v0.9.2 (#2248)
- Seqera Platform CLI: handle failed tasks (#2286)
MultiQC v1.19 - 2023-12-18
- Add missing table
idin DRAGEN modules, and requireidin plot configs in strict mode (#2228) - Config
table_columns_visibleandtable_columns_name: support flat config andtable_idas a group (#2191) - Add
sort_samples: falseconfig option for bar graphs (#2210) - Upgrade the jQuery tablesorter plugin to v2 (#1666)
- Refactor pre-Python-3.6 code, prefer f-strings over
.format()calls (#2224) - Allow specifying default sort columns for tables with
defaultsort(#1667) - Create CODE_OF_CONDUCT.md (#2195)
- Add
.cramto sample name cleaning defaults (#2209)
- Re-add
runinto themultiqcnamespace (#2202) - Fix the
"square": Trueflag to scatter plot to actually make the plot square (#2189) - Fix running with the
--no-reportflag (#2212) - Fix guessing custom content plot type: do not assume first row of a bar plot data are sample names (#2208)
- Fix detection of changed specific module in Changelog CI (#2234)
- BCLConvert: fix mean quality, fix count-per-lane bar plot (#2197)
- deepTools: handle missing data in
plotProfile(#2229) - Fastp: search content instead of file name (#2213)
- GATK: square the
BaseRecalibratorscatter plot (#2189) - HiC-Pro: add missing search patterns and better handling of missing data (#2233)
- Kraken: fix
UnboundLocalError(#2230) - Kraken: fixed column keys in genstats (#2205)
- QualiMap: fix
BamQCfor global-only stats (#2207) - Picard: add more search patterns for
MarkDuplicates, includingMarkDuplicatesSpark(#2226) - Salmon: add
library_types,compatible_fragment_ratio,strand_mapping_biasto the general stats table (#1485)
MultiQC v1.18 - 2023-11-17
As of this release, you can now set all of your config variables via environment variables! (see docs).
Better still, YAML config files can now use string interpolation to parse environment variables within strings (see docs), eg:
report_header_info:
- Contact E-mail: !ENV "${NAME:info}@${DOMAIN:example.com}"In this release, there was a significant refactoring of the Picard module. It has been generalized for better code sharing with other Picard-based software, like Sentieon and Parabricks. As a result of this, the standalone Sentieon module was removed: Sentieon QC files will be interpreted directly as Picard QC files.
If you were using the Sentieon module in your pipelines, make sure to update any places that reference the module name:
- MultiQC command line (e.g. replace
--module sentieonwith--module picard). - MultiQC configs (e.g. replace
sentieonwithpicardin options likerun_modules,exclude_modules,module_order). - Downstream code that relies on names of the files in
multiqc_dataormultiqc_plotssaves (e.g.,multiqc_data/multiqc_sentieon_AlignmentSummaryMetrics.txtbecomesmultiqc_data/multiqc_picard_AlignmentSummaryMetrics.txt). - Code that parses data files like
multiqc_data/multiqc_data.json. - Custom plugins and templates that rely on HTML anchors (e.g.
#sentieon_aligned_readsbecomes#picard_AlignmentSummaryMetrics). - Also, note that Picard fetches sample names from the commands it finds inside the QC headers (e.g.
# net.sf.picard.analysis.CollectMultipleMetrics INPUT=Szabo_160930_SN583_0215_AC9H20ACXX.bam ...->Szabo_160930_SN583_0215_AC9H20ACXX), whereas the removed Sentieon module prioritized the QC file names. To revert to the old Sentieon approach, use theuse_filename_as_sample_nameconfig flag.
- Config can be set with environment variables, including env var interpolation (#2178)
- Try find config in
~/.configor$XDG_CONFIG_HOME(#2183) - Better sample name cleaning with pairs of input filenames (#2181)
- Software versions: allow any string as a version tag (#2166)
- Table columns with non-numeric values and now trigger a linting error if
scaleis set (#2176) - Stricter config variable typing (#2178)
- Remove
position:absoluteCSS from table values (#2169) - Fix column sorting in exported TSV files from a matplotlib linegraph plot (#2143)
- Fix custom anchors for kraken (#2170)
- Fix logging spillover bug (#2174)
- Seqera Platform CLI (#2151)
- Seqera Platform CLI reports statistics generated by the Seqera Platform CLI.
- Xenome (#1860)
- A tool for classifying reads from xenograft sources.
- xengsort (#2168)
- xengsort is a fast xenograft read sorter based on space-efficient k-mer hashing
- fastp: add version parsing (#2159)
- fastp: correctly parse sample name from
--in1/--in2in bash command. Prefer file name if notfastp.json; fallback to file name when error (#2139) - Kaiju: fix
division by zeroerror (#2179) - Nanostat: account for both tab and spaces in
v1.41+search pattern (#2155) - Pangolin: update for v4: add QC Note , update tool versions columns (#2157)
- Picard: Generalize to directly support Sentieon and Parabricks outputs (#2110)
- Sentieon: Removed the module in favour of directly supporting parsing by the Picard module (#2110)
- Note that any code that relies on the module name needs to be updated, e.g.
-m sentieonwill no longer work - The exported plot and data files will be now be prefixed as
picardinstead ofsentieon, etc. - Note that the Sentieon module used to fetch the sample names from the file names by default, and now it follows the Picard module's logic, and prioritizes the commands recorded in the logs. To override, use the
use_filename_as_sample_nameconfig flag
- Note that any code that relies on the module name needs to be updated, e.g.
MultiQC v1.17 - 2023-10-17
Highlights:
- Introducing the new MultiQC logo!
- Adding support for Python 3.12 and dropping support for Python 3.7
- New
--require-logsto fail if expected tool outputs are not found - Rename
--lintto--strict - Modules should now use
ModuleNotFoundErrorinstead ofUserWarningwhen no logs are found - 2 new modules and updates to 9 modules.
- Add CI action changelog.yml to populate the changelog from PR titles, triggered by a comment
@multiqc-bot changelog(#2025, #2102, #2115) - Add GitHub Actions bot workflow to fix code linting from a PR comment (#2082)
- Use custom exception type instead of
UserWarningwhen no samples are found. (#2049) - Lint modules for missing
self.add_software_version(#2081) - Strict mode: rename
config.linttoconfig.strict, crash early on module or template error. AddMULTIQC_STRICT=1(#2101) - Matplotlib line plots now respect
xLog: TrueandyLog: Truein config (#1632) - Fix matplotlib linegraph and bargraph for the case when
xmax<xminin config (#2124) - Add
--require-logsflag to error out if requested modules not used (#2109) - Fixes for python 3.12
- Drop Python 3.6 and 3.7 support, add 3.12 (#2121)
- Just run CI on the oldest + newest supported Python versions (#2074)
 New logo
New logo- Set name and anchor for the custom content "module" #2131
- Fix use of
shutil.copytreewhen overriding existing template files intmp_dir(#2133)

- Bracken
- A highly accurate statistical method that computes the abundance of species in DNA sequences from a metagenomics sample.
- Truvari (#1751)
- Truvari is a toolkit for benchmarking, merging, and annotating structural variants
- Dragen: make sure all inputs are recorded in multiqc_sources.txt (#2128)
- Cellranger: Count submodule updated to parse Antibody Capture summary (#2118)
- fastp: parse unescaped sample names with white spaces (#2108)
- FastQC: Add top overrepresented sequences table (#2075)
- HiCPro: Fix parsing scientific notation in hicpro-ashic. Thanks @Just-Roma (#2126)
- HTSeq Count: allow counts files with more than 2 columns (#2129)
- mosdepth: fix prioritizing region over global information (#2106)
- Picard: Adapt WgsMetrics to parabricks bammetrics outputs (#2127)
- Picard: MarkDuplicates: Fix parsing mixed strings/numbers, account for missing trailing
0(#2083, #2094) - Samtools: Add MQ0 reads to the Percent Mapped barplot in Stats submodule (#2123)
- WhatsHap: Process truncated input with no ALL chromosome (#2095)
MultiQC v1.16 - 2023-09-22
New in v1.16 - software version information can now automatically parsed from log output where available,
and added to MultiQC in a standardised manner. It's shown in the MultiQC report next to section
headings and in a dedicated report section, as well as being saved to multiqc_data.
Where version information is not available in logs, it can be submitted manually by using a new
special file type with filename pattern *_mqc_versions.yml.
There's the option of representing groups of versions, useful for a tool that uses sub-tools,
or pipelines that want to report version numbers per analysis step.
There are a handful of new config scopes to control behaviour:
software_versions, skip_versions_section, disable_version_detection, versions_table_group_header.
See the documentation for more (writing modules,
supplying stand-alone)
Huge thanks to @pontushojer for the contribution (#1927). This idea goes way back to issue #290, made in 2016!
- Removed
simplejsonunused dependency (#1973) - Give config
custom_plot_configpriority over column-specific settings set by modules - When exporting plots, make a more clear error message for unsupported FastQC dot plot (#1976)
- Fixed parsing of
plot_type: "html"datain json custom content - Replace deprecated
pkg_resources - Fix the module groups configuration for modules where the namespace is passed explicitly to
general_stats_addcols. Namespace is now always appended to the module name in the general stats (2037). - Do not call
sys.exit()in themultiqc.run()function, to avoid breaking interactive environments. #2055 - Fixed the DOI exports in
multiqc_datato include more than just the MultiQC paper (#2058) - Fix table column color scaling then there are negative numbers (1869)
- Export plots as static images and data in a ZIP archive. Fixes the issue when only 10 plots maximum were downloaded due to the browser limitation.
- Bakta
- Rapid and standardized annotation of bacterial genomes, MAGs & plasmids.
- mapDamage
- mapDamage2 is a computational framework written in Python and R, which tracks and quantifies DNA damage patterns among ancient DNA sequencing reads generated by Next-Generation Sequencing platforms.
- Sourmash
- Quickly search, compare, and analyze genomic and metagenomic data sets.
- BcfTools
- Stats: fix parsing multi-sample logs (#2052)
- Custom content
- Don't convert sample IDs to floats (#1883)
- DRAGEN
- Make DRAGEN module use
fn_clean_extsinstead of hardcoded file names. Fixes working with arbitrary file names (#1994)
- Make DRAGEN module use
- FastQC:
- fix
UnicodeDecodeErrorwhen parsingfastqc_data.txt: try latin-1 or fail gracefully (#2024)
- fix
- Kaiju:
- Fix
UnboundLocalErroron outputs when Kanju was run with the-eflag (#2023)
- Fix
- Kraken
- Mosdepth
- Add X/Y relative coverage plot, analogous to the one in samtools-idxstats (#1978)
- Added the
perchrom_fraction_cutoffoption into the config to help avoid clutter in contig-level plots - Fix a bug happening when both
regionandglobalcoverage histograms for a sample are available (i.e. when mosdepth was run with--by, see mosdepth docs). In this case, data was effectively merged. Instead, summarise it separately and add a separate report section for the region-based coverage data. - Do not fail when all input samples have no coverage (#2005).
- NanoStat
- Support new format (#1997).
- RSeQC
- Samtools
- Stats: fix "Percent Mapped" plot when samtools was run with read filtering (#1972)
- Qualimap
- BamQC: Include
% On Targetin General Stats table (#2019)
- BamQC: Include
- WhatsHap
- Bugfix: ensure that TSV is only split on tab character. Allows sample names with spaces (#1981)
MultiQC v1.15 - 2023-08-04
This release of MultiQC introduces speed improvements to the file search. One way it does this is by limiting the number of lines loaded by each search pattern. For the vast majority of users, this should have no effect except faster searches. However, in some edge cases it may break things. Hypothetically, for example:
- If you concatenate log files from multiple tools
- If you have a custom plugin module that we haven't tested
See the troubleshooting docs for more information.
- Refactor file search for performance improvements (#1904)
- Bump
log_filesize_limitdefault (to skip large files in the search) from 10MB to 50MB. - Table code now tolerates lambda function calls with bad data (#1739)
- Beeswarm plot now saves data to
multiqc_data, same as tables (#1861) - Don't print DOI in module if it's set to an empty string.
- Optimize parsing of 2D data dictionaries in multiqc.utils.utils_functions.write_data_file() (#1891)
- Don't sort table headers alphabetically if we don't have an
OrderedDict- regular dicts are fine in Py3 (#1866) - New back-end to preview + deploy the new website when the docs are edited.
- Fixed a lot of broken links in the documentation from the new website change in structure.
- Freyja
- Freyja is a tool to recover relative lineage abundances from mixed SARS-CoV-2 samples from a sequencing dataset.
- Librarian
- A tool to predict the sequencing library type from the base composition of a supplied FastQ file.
- Conpair
- Bugfix: allow to find and proprely parse the
concordanceoutput of Conpair, which may output 2 kinds of format forconcordancedepending if it's ran with or without--outfile(#1851)
- Bugfix: allow to find and proprely parse the
- Cell Ranger
- DRAGEN
- filtlong
- Handle reports from locales that use
.as a thousands separator (#1843)
- Handle reports from locales that use
- GATK
- Adds support for AnalyzeSaturationMutagenesis submodule
- HUMID
- Fix bug that prevent HUMID stats files from being parsed (#1856)
- Mosdepth
- Fix data not written to
mosdepth_cumcov_dist.txtandmosdepth_cov_dist.txt(#1868) - Update documentation with new file
{prefix}.mosdepth.summary.txt(#1868) - Fill in missing values for general stats table (#1868)
- Include mosdepth/summary file paths in
multiqc_sources.txt(#1868) - Enable log switch for Coverage per contig plot (#1868)
- Fix y-axis scaling for Coverage distribution plot (#1868)
- Handle case of intermediate missing coverage x-values in the
*_dist.txtfile causing a distorted Coverage distribution plot (#1960)
- Fix data not written to
- Picard
- Porechop
- Don't render bar graphs if no samples had any adapters trimmed (#1850)
- Added report section listing samples that had no adapters trimmed
- RSeQC
- Fix
ZeroDivisionErrorerror forbam_statresults when there are 0 reads (#1735)
- Fix
- UMI-tools
- Fix bug that broke the module with paired-end data (#1845)
MultiQC v1.14 - 2023-01-08
- Rewrote the
Dockerfileto build multi-arch images (amd64 + arm), run through a non-privileged user and build tools for non precompiled python binaries (#1541, #1541) - Add a new lint test to check that colour scale names are valid (#1835)
- Update github actions to run tests on a single module if it is the only file affected by the PR (#915)
- Add CI testing for Python 3.10 and 3.11
- Optimize line-graph generation to remove an n^2 loop (#1668)
- Parsing output file column headers is much faster.
- Remove Python 2-3 compatability
from __future__imports - Remove unused
#!/usr/bin/env pythonhashbangs from module files - Add new code formatting tool isort to standardise the order and formatting of Python module imports
- Add Pycln pre-commit hook to remove unused imports
- Bugfix: Make
config.data_formatwork again (#1722) - Bump minimum version of Jinja2 to
>=3.0.0(#1642) - Disable search progress bar if running with
--quietor--no-ansi(#1638) - Allow path filters without full paths by trying to prefix analysis dir when filtering (#1308)
- Fix sorting of table columns with text values
- Don't crash if a barplot is given an empty list of categories (#1540)
- New logos! MultiQC is now developed and maintained at Seqera Labs. Updated logos and email addresses accordingly.
- Anglerfish
- A tool designed to assess pool balancing, contamination and insert sizes of Illumina library dry runs on Oxford Nanopore data.
- BBDuk
- Combines most common data-quality-related trimming, filtering, and masking operations via kmers into a single high-performance tool.
- Cell Ranger
- Works with data from 10X Genomics Chromium. Processes Chromium single cell data to align reads, generate feature-barcode matrices, perform clustering and other secondary analysis, and more.
- New MultiQC module parses Cell Ranger quality reports from VDJ and count analysis
- DIAMOND
- A high-throughput program for aligning DNA reads or protein sequences against a protein reference database.
- DRAGEN-FastQC
- Illumina Bio-IT Platform that uses FPGA for accelerated primary and secondary analysis
- Finally merged the epic 2.5-year-old pull request, with 3.5k new lines of code.
- Please report any bugs you find!
- Filtlong
- A tool for filtering long reads by quality.
- GoPeaks
- GoPeaks is used to call peaks in CUT&TAG/CUT&RUN datasets.
- HiFiasm
- A haplotype-resolved assembler for accurate Hifi reads
- HUMID
- HUMID is a tool to quickly and easily remove duplicate reads from FastQ files, with or without UMIs.
- mOTUs
- Microbial profiling through marker gene (MG)-based operational taxonomic units (mOTUs)
- Nextclade
- Tool that assigns clades to SARS-CoV-2 samples
- Porechop
- A tool for finding and removing adapters from Oxford Nanopore reads
- PRINSEQ++
- PRINSEQ++ is a C++ of
prinseq-lite.plprogram for filtering, reformating or trimming genomic and metagenomic sequence data.
- PRINSEQ++ is a C++ of
- UMI-tools
- Work with Unique Molecular Identifiers (UMIs) / Random Molecular Tags (RMTs) and single cell RNA-Seq cell barcodes.
- Bcftools stats
- BCL Convert
- Handle single-end read data correctly when setting cluster length instead of always assuming paired-end reads (#1697)
- Handle different R1 and R2 read-lengths correctly instead of assuming they are the same (#1774)
- Handle single-index paired-end data correctly
- Added a config option to enable the creation of barplots with undetermined barcodes (
create_unknown_barcode_barplotswithFalseas default) (#1709)
- BUSCO
- Update BUSCO pass/warning/fail scheme to be more clear for users
- Bustools
- Show median reads per barcode statistic
- Custom content
- fastp
- FastQC
- Report median read-length for fastqc in addition to mean (#1745)
- Kaiju
- Don't crash if we don't have any data for the top-5 barplot (#1540)
- Kallisto
- Fix
ZeroDivisionErrorwhen a sample has zero reads (#1746)
- Fix
- Kraken
- malt
- Fixed division by 0 in malt module (#1683)
- miRTop
- Avoid
KeyError- don't assume all fields present in logs (#1778)
- Avoid
- Mosdepth
- Don't pad the General Stats table with zeros for missing data (#1810)
- Picard
- HsMetrics: Allow custom columns in General Stats too, with
HsMetrics_genstats_table_colsandHsMetrics_genstats_table_cols_hidden
- HsMetrics: Allow custom columns in General Stats too, with
- Qualimap
- RSeQC
- Update
geneBody_coverageto plot normalized coverages using a similar formula to that used by RSeQC itself (#1792)
- Update
- Sambamba Markdup
- Catch zero division in sambamba markdup (#1654)
- Samtools
- Added additional column for
flagstatthat displays percentage of mapped reads in a bam (hidden by default) (#1733)
- Added additional column for
- VEP
- Don't crash with
ValueErrorif there are zero variants (#1681)
- Don't crash with
MultiQC v1.13 - 2022-09-08
- Major spruce of the command line help, using the new rich-click package
- Drop some of the Python 2k compatability code (eg. custom requirements)
- Improvements for running MultiQC in a Python environment, such as a Jupyter Notebook or script
- Fixed bug raised when removing logging file handlers between calls that arose when configuring the root logger with dictConfig (#1643)
- Added new config option
custom_table_header_configto override any config for any table header - Fixed edge-case bug in custom content where a
descriptionthat doesn't terminate in.gave duplicate section descriptions. - Tidied the verbose log to remove some very noisy statements and add summaries for skipped files in the search
- Add timezone to time in reports
- Add nix flake support
- Added automatic tweet about new releases
- Breaking: Removed
--cl_configoption. Please use--cl-configinstead.
- AdapterRemoval
- Finally merge a fix for counts of reads that are discarded/collapsed (#1647)
- VEP
- Fixed bug when
General Statisticshave a value of-(#1656)
- Fixed bug when
- Custom content
- Nanostat
- Pangolin
- Updated module to handle outputs from Pangolin v4 (#1660)
- Somalier
- Handle zero mean X depth in Sex plot (#1670)
- Fastp
- Include low complexity and too long reads in filtering bar chart
- miRTop
- FastQC
- Fixed error when parsing duplicate ratio when there is
nanvalues in the report. (#1725)
- Fixed error when parsing duplicate ratio when there is
MultiQC v1.12 - 2022-02-08
- Added option to customise default plot height in plot config (#1432)
- Added
--no-reportflag to skip report generation (#1462) - Added support for priting tool DOI in report sections (#1177)
- Added support for
--custom-css-file/config.custom_css_filesoption to include custom CSS in the final report (#1573) - New plot config option
labelSizeto customise font size for axis labels in flat MatPlotLib charts (#1576) - Added support for customising table column names (#1255)
- MultiQC now skips modules for which no files were found - gives a small performance boost (#1463)
- Improvements for running MultiQC in a Python environment, such as a Jupyter Notebook or script
- Added commonly missing functions to several modules (#1468)
- Wrote new script to check for the above function calls that should be in every module (
.github/workflows/code_checks.py), runs on GitHub actions CI - Make table Conditional Formatting work at table level as well as column level. (#761)
- CSS Improvements to make printed reports more attractive / readable (#1579)
- Fixed a problem with numeric filenames (#1606)
- Fixed nasty bug where line charts with a categorical x-axis would take categories from the last sample only (#1568)
- Ignore any files called
multiqc_data.json(#1598) - Check that the config
path_filtersis a list, convert to list if a string is supplied (#1539)
- CheckQC
- A program designed to check a set of quality criteria against an Illumina runfolder
- pbmarkdup
- Mark duplicate reads from PacBio sequencing of an amplified library
- WhatsHap
- WhatsHap is a software for phasing genomic variants using DNA sequencing reads
- SeqWho
- Tool to determine a FASTQ(A) sequencing file identity, both source protocol and species of origin.
- BBMap
- Added handling for
qchistoutput (#1021)
- Added handling for
- bcftools
- Added a plot with samplewise number of sites, Ts/Tv, number of singletons and sequencing depth (#1087)
- Mosdepth
- Added mean coverage #1566
- NanoStat
- Recognize FASTA and FastQ report flavors (#1547)
- BBMap
- Correctly handle adapter stats files with additional columns (#1556)
- BCL Convert
- Handle change in output format in v3.9.3 with new
Quality_Metrics.csvfile (#1563)
- Handle change in output format in v3.9.3 with new
- bowtie
- Minor update to handle new log wording in bowtie v1.3.0 (#1615)
- CCS
- Custom content
- DRAGEN
- Fixed bug in sample name regular expression (#1537)
- Fastp
- Fixed % pass filter statistics (#1574)
- FastQC
- goleft/indexcov
- Fix
ZeroDivisionErrorif no bins are found (#1586)
- Fix
- HiCPro
- Better handling of errors when expected data keys are not found (#1366)
- Lima
- Move samples that have been renamed using
--replace-namesinto the General Statistics table (#1483)
- Move samples that have been renamed using
- miRTrace
- Replace hardcoded RGB colours with Hex to avoid errors with newer versions of matplotlib (#1263)
- Mosdepth
- Fixed issue #1568
- Fixed a bug when reporting per contig coverage
- Picard
- Update
ExtractIlluminaBarcodesto recognise more log patterns in newer versions of Picard (#1611)
- Update
- Qualimap
- Fix
ZeroDivisionErrorinQM_RNASeqand skip genomic origins plot if no aligned reads are found (#1492)
- Fix
- QUAST
- Clarify general statistics table header for length
- RSeQC
- Sambamba
- Fixed issue with a change in the format of output from
sambamba markdup0.8.1 (#1617)
- Fixed issue with a change in the format of output from
- Skewer
- Fix
ZeroDivisionErrorif no available reads are found (#1622)
- Fix
- Somalier
- Plot scaled X depth instead of mean for Sex plot (#1546)
- VEP
- Handle table cells containing
-instead of numbers (#1597)
- Handle table cells containing
MultiQC v1.11 - 2021-07-05
- New interactive slider controls for controlling heatmap colour scales (#1427)
- Added new
--replace-names/ configsample_names_replaceoption to replace sample names during report generation - Added
use_filename_as_sample_nameconfig option /--fn_as_s_namecommand line flag (#949, #890, #864, #998, #1390)- Forces modules to use the log filename for the sample identifier, even if the module usually takes this from the file contents
- Required a change to the
clean_s_name()function arguments. All core MultiQC modules updated to reflect this. - Should be backwards compatible for custom modules. To adopt new behaviour, supply
finstead off["root"]as the second argument. - See the documenation for details: Using log filenames as sample names and Custom sample names.
- Make the module crash tracebacks much prettier using
rich - Refine the cli log output a little (nicely formatted header line + drop the
[INFO]) - Added docs describing tools for downstream analysis of MultiQC outputs.
- Added CI tests for Python 3.9, pinned
networkxpackage to>=2.5.1(#1413) - Added patterns to
config.fn_ignore_pathsto avoid error with parsing installation dir / singularity cache (#1416) - Print a log message when flat-image plots are used due to sample size surpassing
plots_flat_numseriesconfig (#1254) - Fix the
mqc_coloursutil function to lighten colours even when passing categorical or single-length lists. - Bugfix for Custom Content, using YAML configuration (eg. section headers) for images should now work
- BCL Convert
- Tool that converts / demultiplexes Illumina Binary Base Call (BCL) files to FASTQ files
- Bustools
- Tools for working with BUS files
- ccs
- Generate highly accurate single-molecule consensus reads from PacBio data
- GffCompare
- GffCompare can annotate and estimate accuracy of one or more GFF files compared with a reference annotation.
- Lima
- The PacBio Barcode Demultiplexer
- NanoStat
- Calculate various statistics from a long read sequencing datasets
- ODGI
- Optimized dynamic genome/graph implementation
- Pangolin
- Added MultiQC support for Pangolin, the tool that determines SARS-CoV-2 lineages
- Sambamba Markdup
- Added MultiQC module to add duplicate rate calculated by Sambamba Markdup.
- Snippy
- Rapid haploid variant calling and core genome alignment.
- VEP
- Added MultiQC module to add summary statistics of Ensembl VEP annotations.
- Handle error from missing variants in VEP stats file. (#1446)
- Cutadapt
- Added support for linked adapters #1329]
- Parse whether trimming was 5' or 3' for Lengths of Trimmed Sequences plot where possible
- Mosdepth
- Include or exclude contigs based on patterns for coverage-per-contig plots
- Picard
- Add support for
CollectIlluminaBasecallingMetrics,CollectIlluminaLaneMetrics,ExtractIlluminaBarcodesandMarkIlluminaAdapters(#1336) - New
insertsize_xmaxconfiguration option to limit the plotted maximum insert size forInsertSizeMetrics
- Add support for
- Qualimap
- Added new percentage coverage plot in
QM_RNASeq(#1258)
- Added new percentage coverage plot in
- RSeQC
- biscuit
- Duplicate Rate and Cytosine Retention tables are now bargraphs.
- Refactor code to only calculate alignment statistics once.
- Fixed bug where cytosine retentions values would not be properly read if in scientific notation.
- bcl2fastq
- Added sample name cleaning so that prepending directories with the
-dflag works properly.
- Added sample name cleaning so that prepending directories with the
- Cutadapt
- Dragen
- Handled MultiQC crashing when run on single-end output from Dragen (#1374)
- fastp
- Handle a
ZeroDivisionErrorif there are zero reads (#1444)
- Handle a
- FastQC
- Added check for if
overrepresented_sequencesis missing from reports (#1281)
- Added check for if
- Flexbar
- Fixed bug where reports with 0 reads would crash MultiQC (#1407)
- Kraken
- Mosdepth
- Show barplot instead of line graph for coverage-per-contig plot if there is only one contig.
- Picard
RnaSeqMetrics- fix assignment barplot labels to say bases instead of reads (#1408)CrosscheckFingerprints- fix bug where LOD threshold was not detected when invoked with "new" picard cli style. fixed formatting bug (#1414)- Made checker for comma as decimal separator in
HsMetricsmore robust (#1296)
- qc3C
- Updated module to not fail on older field names.
- Qualimap
- Fixed wrong units in tool tip label (#1258)
- QUAST
- Fixed typo causing wrong number of contigs being displayed (#1442)
- Sentieon
- Handled
ZeroDivisionErrorwhen input files have zero reads (#1420)
- Handled
- RSEM
- Handled
ZeroDivisionErrorwhen input files have zero reads (#1040)
- Handled
- RSeQC
- Fixed double counting of some categories in
read_distributionbar graph. (#1457)
- Fixed double counting of some categories in
MultiQC v1.10.1 - 2021-04-01
- Dropped the
Skipping search patternlog message from a warning to debug - Moved directory prepending with
-dback to before sample name cleaning (as it was before v1.7) (#1264) - If linegraph plot data goes above
ymax, only discard the data if the line doesn't come back again (#1257) - Allow scientific notation numbers in colour scheme generation
- Fixed bug with very small minimum numbers that only revelead itself after a bugfix done in the v1.10 release
- Allow
top_modulesto be specified as empty dicts (#1274) - Require at least
richversion9.4.0to avoidSpinnerColumnAttributeError(#1393) - Properly ignore
.snakemakefolders as intended (#1395)
- bcftools
- Fixed bug where
QUALvalue.would crash MultiQC (#1400)
- Fixed bug where
- bowtie2
- Fix bug where HiSAT2 paired-end bar plots were missing unaligned reads (#1230)
- Deeptools
- FastQC
- Replace
NaNwith0in the Per Base Sequence Content plot to avoid crashing the plot (#1246)
- Replace
- Picard
- Fixed bug in
ValidateSamFilemodule where additional whitespace at the end of the file would cause MultiQC to crash (#1397)
- Fixed bug in
- Somalier
- Fixed bug where using sample name cleaning in a config would trigger a
KeyError(#1234)
- Fixed bug where using sample name cleaning in a config would trigger a
MultiQC v1.10 - 2021-03-08
This is a big change for MultiQC developers. I have added automated code formatting and code linting (style checks) to MultiQC. This helps to keep the MultiQC code base consistent despite having many contributors and helps me to review pull-requests without having to consider whitespace.
Specifically, MultiQC now uses three main tools:
- Black - Python Code
- Prettier - Everything else (almost)
- markdownlint-cli - Stricter markdown rules
All developers must run these tools when submitting changes via Pull-Requests! Automated CI tests now run with GitHub actions to check that all files pass the above tests. If any files do not, that test will fail giving a red ❌ next to the pull request.
For further information, please see the documentation.
--sample-filtersnow also acceptsshow_reandhide_rein addition toshowandhide. The_reoptions use regex, while the "normal" options use globbing.- MultiQC config files now work with
.ymlfile extension as well as.yaml.yamlwill take preference if both found.
- Section comments can now also be added for General Statistics
section_comments: { general_stats: "My comment" }
- New table header config option
bgcolsallows background colours for table cells with categorical data. - New table header config options
cond_formatting_rulesandcond_formatting_colours- Comparable functionality to user config options
table_cond_formatting_rulesandtable_cond_formatting_colours, allowes module developers to format table cell values as labels.
- Comparable functionality to user config options
- New CI test looks for git merge markers in files
- Beautiful new progress bar from the amazing willmcgugan/rich package.
- Added a bunch of new default sample name trimming suffixes (see
8ac5c7b) - Added
timeout-minutes: 10to the CI test workflow to check that changes aren't negatively affecting run time too much. - New table header option
bars_zero_centrepointto treat0as zero width bars and plot bar length based on absolute values
- EigenStratDatabaseTools
- Added MultiQC module to report SNP coverages from
eigenstrat_snp_coverage.pyin the general stats table.
- Added MultiQC module to report SNP coverages from
- HOPS
- Post-alignment ancient DNA analysis tool for MALT
- JCVI
- Computes statistics on genome annotation.
- ngsderive
- Forensic analysis tool useful in backwards computing information from next-generation sequencing data.
- OptiType
- Precision HLA typing from next-generation sequencing data
- PURPLE
- A purity, ploidy and copy number estimator for whole genome tumor data
- Pychopper
- Identify, orient and trim full length Nanopore cDNA reads
- qc3C
- Reference-free QC of Hi-C sequencing data
- Sentieon
- Submodules added to catch Picard-based QC metrics files
- DRAGEN
- featureCounts
- fgbio
- Fix
ErrorRateByReadPositionto calculateymaxnot just on the overallerror_rate, but also specific base errors (ex.a_to_c_error_rate,a_to_g_error_rate, ...). (#1215) - Fix
ErrorRateByReadPositionplotted line names to no longer concatenate multiple read identifiers and no longer have off-by-one read numbering (ex.Sample1_R2_R3->Sample1_R2) ([#1304)
- Fix
- Fastp
- Fixed description for duplication rate (pre-filtering, not post) ([#1313)
- GATK
- Add support for the creation of a "Reported vs Empirical Quality" graph to the Base Recalibration module.
- hap.py
- Updated module to plot both SNP and INDEL stats (#1241)
- indexcov
- Fixed bug when making the PED file plots (#1265)
- interop
- Added the
% Occupiedmetric toRead Metrics per Lanetable which is reported for NovaSeq and iSeq platforms.
- Added the
- Kaiju
- Kraken
- MALT
- Fix y-axis labelling in bargraphs
- MACS2
- Add number of peaks to the General Statistics table.
- mosdepth
- Enable prepending of directory to sample names
- Display contig names in Coverage per contig plot tooltip
- Picard
- Fix
HsMetricsbait percentage columns (#1212) - Fix
ConvertSequencingArtifactToOxoGfiles not being found (#1310) - Make
WgsMetricshistogram smoothed if more than 1000 data points (avoids huge plots that crash the browser) - Multiple new config options for
WgsMetricsto customise coverage histogram and speed up MultiQC with very high coverage files. - Add additional datasets to Picard Alignment Summary (#1293)
- Add support for
CrosscheckFingerprints(#1327)
- Fix
- PycoQC
- Log10 x-axis for Read Length plot (#1214)
- Rockhopper
- Fix issue with parsing genome names in Rockhopper summary files (#1333)
- Fix issue properly parsing multiple samples within a single Rockhopper summary file
- Salmon
- Only try to generate a plot for fragment length if the data was found.
- verifyBamID
- Fix
CHIPvalue detection (#1316).
- Fix
- General Stats custom content now gives a log message
- If
idis not set inJSONorYAMLfiles, it defaults to the sample name instead of justcustom_content - Data from
JSONorYAMLnow hasdatakeys (sample names) run through theclean_s_name()function to apply sample name cleanup - Fixed minor bug which caused custom content YAML files with a string
datatype to not be parsed
- Disable preservation of timestamps / modes when copying temp report files, to help issues with network shares (#1333)
- Fixed MatPlotLib warning:
FixedFormatter should only be used together with FixedLocator - Fixed long-standing min/max bug with shared minimum values for table columns using
shared_key - Made table colour schemes work with negative numbers (don't strip
-from values when making scheme)
MultiQC v1.9 - 2020-05-30
Python 2 had its official sunset date on January 1st 2020, meaning that it will no longer be developed by the Python community. Part of the python.org statement reads:
That means that we will not improve it anymore after that day, even if someone finds a security problem in it. You should upgrade to Python 3 as soon as you can.
Very many Python packages no longer support Python 2 and it whilst the MultiQC code is currently compatible with both Python 2 and Python 3, it is increasingly difficult to maintain compatibility with the dependency packages it uses, such as MatPlotLib, numpy and more.
As of MultiQC version 1.9, Python 2 is no longer officially supported. Automatic CI tests will no longer run with Python 2 and Python 2 specific workarounds are no longer guaranteed.
Whilst it may be possible to continue using MultiQC with Python 2 for a short time by pinning dependencies, MultiQC compatibility for Python 2 will now slowly drift and start to break. If you haven't already, you need to switch to Python 3 now.
- Now using GitHub Actions for all CI testing
- Dropped Travis and AppVeyor, everything is now just on GitHub
- Still testing on both Linux and Windows, with multiple versions of Python
- CI tests should now run automatically for anyone who forks the MultiQC repository
- Linting with
--lintnow checks line graphs as well as bar graphs - New
gatheredtemplate with no tool name sections (#1119) - Added
--sample-filtersoption to add show/hide buttons at the top of the report (#1125)- Buttons control the report toolbox Show/Hide tool, filtering your samples
- Allows reports to be pre-configured based on a supplied list of sample names at report-generation time.
- Line graphs can now have
Log10buttons (same functionality as bar graphs) - Importing and running
multiqcin a script is now a little Bettermultiqc.runnow returns thereportandconfigas well as the exit code. This means that you can explore the MultiQC run time a little in the Python environment.- Much more refactoring is needed to make MultiQC as useful in Python scripts as it could be. Watch this space.
- If a custom module
anchoris set usingmodule_order, it's now used a bit more:- Prefixed to module section IDs
- Appended to files saved in
multiqc_data - Should help to prevent duplicates requiring
-1suffixes when running a module multiple times
- New heatmap plot config options
xcats_samplesandycats_samples- If set to
False, the report toolbox options (highlight, rename, show/hide) do not affect that axis. - Means that the Show only matching samples report toolbox option works on FastQC Status Checks, for example (#1172)
- If set to
- Report header time and analysis paths can now be hidden
- New config options
show_analysis_pathsandshow_analysis_time(#1113)
- New config options
- New search pattern key
skip: trueto skip specific searches when modules look for a lot of different files (eg. Picard). - New
--profile-runtimecommand line option (config.profile_runtime) to give analysis of how long the report takes to be generated- Plots of the file search results and durations are added to the end of the MultiQC report as a special module called Run Time
- A summary of the time taken for the major stages of MultiQC execution are printed to the command line log.
- New table config option
only_defined_headers- Defaults to
true, set tofalseto also show any data columns that are not defined as headers - Useful as allows table-wide defaults to be set with column-specific overrides
- Defaults to
- New
modulekey allowed forconfig.extra_fn_clean_extsandconfig.fn_clean_exts- Means you can limit the action of a sample name cleaning pattern to specific MultiQC modules (#905)
- Improve support for HTML files - now just end your HTML filename with
_mqc.html- Native handling of HTML snippets as files, no MultiQC config or YAML file required.
- Also with embedded custom content configuration at the start of the file as a HTML comment.
- Add ability to group custom-content files into report sections
- Use the new
parent_id,parent_nameandparent_descriptionconfig keys to group content together like a regular module (#1008)
- Use the new
- Custom Content files can now be configured using
custom_data, without giving search patterns or data
- DRAGEN
- Illumina Bio-IT Platform that uses FPGA for secondary NGS analysis
- iVar
- Added support for iVar: a computational package that contains functions broadly useful for viral amplicon-based sequencing.
- Kaiju
- Fast and sensitive taxonomic classification for metagenomics
- Kraken
- K-mer matching tool for taxonomic classification. Module plots bargraph of counts for top-5 hits across each taxa rank. General stats summary.
- MALT
- Megan Alignment Tool: Metagenomics alignment tool.
- miRTop
- Command line tool to annotate miRNAs with a standard mirna/isomir naming (mirGFF3)
- Module started by @oneillkza and completed by @FlorianThibord
- MultiVCFAnalyzer
- Combining multiple VCF files into one coherent report and format for downstream analysis.
- Picard - new submodules for
QualityByCycleMetrics,QualityScoreDistributionMetrics&QualityYieldMetrics- See #1116
- Rockhopper
- RNA-seq tool for bacteria, includes bar plot showing where features map.
- Sickle
- A windowed adaptive trimming tool for FASTQ files using quality
- Somalier
- Relatedness checking and QC for BAM/CRAM/VCF for cancer, DNA, BS-Seq, exome, etc.
- VarScan2
- Variant calling and somatic mutation/CNV detection for next-generation sequencing data
- BISCUIT
- Major rewrite to work with new BISCUIT QC script (BISCUIT
v0.3.16+)- This change breaks backwards-compatability with previous BISCUIT versions. If you are unable to upgrade BISCUIT, please use MultiQC v1.8.
- Fixed error when missing data in log files (#1101)
- Major rewrite to work with new BISCUIT QC script (BISCUIT
- bcl2fastq
- Samples with multiple library preps (i.e barcodes) will now be handled correctly (#1094)
- BUSCO
- Updated log search pattern to match new format in v4 with auto-lineage detection option (#1163)
- Cutadapt
- New bar plot showing the proportion of reads filtered out for different criteria (eg. too short, too many Ns) (#1198)
- DamageProfiler
- Removes redundant typo in init name. This makes referring to the module's column consistent with other modules when customising general stats table.
- DeDup
- Updates plots to make compatible with 0.12.6
- Fixes reporting errors - barplot total represents mapped reads, not total reads in BAM file
- New: Adds 'Post-DeDup Mapped Reads' column to general stats table.
- FastQC
- FastQ Screen
- fgbio
- New: Plot error rate by read position from
ErrorRateByReadPosition - GroupReadsByUmi plot can now be toggled to show relative percents (#1147)
- New: Plot error rate by read position from
- FLASh
- Logs not reporting innie and outine uncombined pairs now plot combined pairs instead (#1173)
- GATK
- Made parsing for VariantEval more tolerant, so that it will work with output from the tool when run in different modes (#1158)
- MTNucRatioCalculator
- Fixed misleading value suffix in general stats table
- Picard MarkDuplicates
- Major change - previously, if multiple libraries (read-groups) were found then only the first would be used and all others ignored. Now, values from all libraries are merged and
PERCENT_DUPLICATIONandESTIMATED_LIBRARY_SIZEare recalculated. Libraries can be kept as separate samples with a new MultiQC configuration option -picard_config: markdups_merge_multiple_libraries: False - Major change - Updated
MarkDuplicatesbar plot to double the read-pair counts, so that the numbers stack correctly. (#1142)
- Major change - previously, if multiple libraries (read-groups) were found then only the first would be used and all others ignored. Now, values from all libraries are merged and
- Picard HsMetrics
- Picard WgsMetrics
- Updated parsing code to recognise new java class string (#1114)
- QualiMap
- RSeqC
- RNASeQC2
- Updated to handle the parsing metric files from the newer rewrite of RNA-SeqQC.
- Samblaster
- Improved parsing to handle variable whitespace (#1176)
- Samtools
- Removes hardcoding of general stats column names. This allows column names to indicate when a module has been run twice (MultiQC#1076).
- Added an observed over expected read count plot for
idxstats(#1118) - Added additional (by default hidden) column for
flagstatthat displays number total number of reads in a bam
- sortmerna
- Fix the bug for the latest sortmerna version 4.2.0 (#1121)
- sexdeterrmine
- Added a scatter plot of relative X- vs Y-coverage to the generated report.
- VerifyBAMID
- Allow files with column header
FREEMIX(alpha)(#1112)
- Allow files with column header
- Added a new test to check that modules work correctly with
--ignore-samples. A lot of them didn't:Mosdepth,conpair,Qualimap BamQC,RNA-SeQC,GATK BaseRecalibrator,SNPsplit,SeqyClean,Jellyfish,hap.py,HOMER,BBMap,DeepTools,HiCExplorer,pycoQC,interop- These modules have now all been fixed and
--ignore-samplesshould work as you expect for whatever data you have.
- Removed use of
shutil.copyto avoid problems with working on multiple filesystems (#1130) - Made folder naming behaviour of
multiqc_plotsconsistent withmultiqc_data- Incremental numeric suffixes now added if folder already exists
- Plots folder properly renamed if using
-n/--filename
- Heatmap plotting function is now compatible with MultiQC toolbox
hideandhighlight(#1136) - Plot config
logswitch_activenow works as advertised - When running MultiQC modules several times, multiple data files are now created instead of overwriting one another (#1175)
- Fixed minor bug where tables could report negative numbers of columns in their header text
- Fixed bug where numeric custom content sample names could trigger a
TypeError(#1091) - Fixed custom content bug HTML data in a config file would trigger a
ValueError(#1071) - Replaced deprecated 'warn()' with 'warning()' of the logging module
- Custom content now supports
section_extraconfig key to add custom HTML after description. - Barplots with
ymaxset now ignore this when you click the Percentages tab.
MultiQC v1.8 - 2019-11-20
- fgbio
- Process family size count hist data from
GroupReadsByUmi
- Process family size count hist data from
- biobambam2
- Added submodule for
bamsormaduptool - Totally cheating - it uses Picard MarkDuplicates but with a custom search pattern and naming
- Added submodule for
- SeqyClean
- Adds analysis for seqyclean files
- mtnucratio
- Added little helper tool to compute mt to nuclear ratios for NGS data.
- mosdepth
- fast BAM/CRAM depth calculation for WGS, exome, or targeted sequencing
- SexDetErrmine
- Relative coverage and error rate of X and Y chromosomes
- SNPsplit
- Allele-specific alignment sorting
- bcl2fastq
- Added handling of demultiplexing of more than 2 reads
- Allow bcl2fastq to parse undetermined barcode information in situations when lane indexes do not start at 1
- BBMap
- Support for scafstats output marked as not yet implemented in docs
- DeDup
- Added handling clusterfactor and JSON logfiles
- damageprofiler
- Added writing metrics to data output file.
- DeepTools
- fastp
- Fix faulty column handling for the after filtering Q30 rate (#936)
- FastQC
- When including a FastQC section multiple times in one report, the Per Base Sequence Content heatmaps now behave as you would expect.
- Added heatmap showing FastQC status checks for every section report across all samples
- Made sequence content individual plots work after samples have been renamed (#777)
- Highlighting samples from status - respect chosen highlight colour in the toolbox (#742)
- FastQ Screen
- When including a FastQ Screen section multiple times in one report, the plots now behave as you would expect.
- Fixed MultiQC linting errors
- fgbio
- Support the new output format of
ErrorRateByReadPositionfirst introduced in version1.3.0, as well as the old output format.
- Support the new output format of
- GATK
- Refactored BaseRecalibrator code to be more consistent with MultiQC Python style
- Handle zero count errors in BaseRecalibrator
- HiC Explorer
- Fixed bug where module tries to parse
QC_table.txt, a new log file in hicexplorer v2.2. - Updated the format of the report to fits the changes which have been applied to the QC report of hicexplorer v3.3
- Updated code to save parsed results to
multiqc_data
- Fixed bug where module tries to parse
- HTSeq
- Fixed bug where module would crash if a sample had zero reads (#1006)
- LongRanger
- Added support for the LongRanger Align pipeline.
- miRTrace
- Fixed bug where a sample in some plots was missed. (#932)
- Peddy
- Fixed bug where sample name cleaning could lead to error. (#1024)
- All plots (including Het Check and Sex Check) now hidden if no data
- Picard
- Modified
OxoGMetricsso that it will find files created with GATKCollectMultipleMetricsandConvertSequencingArtifactToOxoG.
- Modified
- QoRTs
- Fixed bug where
--dirsbroke certain input files. (#821)
- Fixed bug where
- Qualimap
- Added in mean coverage computation for general statistics report
- Creates now tables of collected data in
multiqc_data
- RNA-SeQC
- Updated broken URL link
- RSeQC
- Fixed bug where Junction Saturation plot when clicking a single sample was mislabelling the lines.
- When including a RSeQC section multiple times in one report, clicking Junction Saturation plot now behaves as you would expect.
- Fixed bug where exported data in
multiqc_rseqc_read_distribution.txtfiles had incorrect values for_kbfields (#1017)
- Samtools
- Utilize in-built
read_count_multiplierfunctionality to plotflagstatresults more nicely
- Utilize in-built
- SnpEff
- Increased the default summary csv file-size limit from 1MB to 5MB.
- Stacks
- Fixed bug where multi-population sum stats are parsed correctly (#906)
- TopHat
- Fixed bug where TopHat would try to run with files from Bowtie2 or HiSAT2 and crash
- VCFTools
- Fixed a bug where
tstv_by_qual.pyproduced invalid json from infinity-values.
- Fixed a bug where
- snpEff
- Added plot of effects
- Added some installation docs for windows
- Added some docs about using MultiQC in bioinformatics pipelines
- Rewrote Docker image
- New base image
czentye/matplotlib-minimalreduces image size from ~200MB to ~80MB - Proper installation method ensures latest version of the code
- New entrypoint allows easier command-line usage
- New base image
- Support opening MultiQC on websites with CSP
script-src 'self'with some sha256 exceptions- Plot data is no longer intertwined with javascript code so hashes stay the same
- Made
config.report_section_orderwork for module sub-sections as well as just modules. - New config options
exclude_modulesandrun_modulesto complement-eand-mcli flags. - Command line output is now coloured by default 🌈 (use
--no-ansito turn this off) - Better launch comparability due to code refactoring by @KerstenBreuer and @ewels
- Windows support for base
multiqccommand - Support for running as a python module:
python -m multiqc . - Support for running within a script:
import multiqcandmultiqc.run('/path/to/files')
- Windows support for base
- Config option
custom_plot_confignow works for bargraph category configs as well (#1044) - Config
table_columns_visiblecan now be given a module namespace and it will hide all columns from that module (#541)
- MultiQC now ignores all
.md5files - Use
SafeLoaderfor PyYaml load calls, avoiding recent warning messages. - Hide
multiqc_config_example.yamlin thetestdirectory to stop people from using it without modification. - Fixed matplotlib background colour issue (@epakarin - #886)
- Table rows that are empty due to hidden columns are now properly hidden on page load (#835)
- Sample name cleaning: All sample names are now truncated to their basename, without a path.
- This includes for
regexandreplace(before was only the defaulttruncate). - Only affects modules that take sample names from file contents, such as cutadapt.
- See #897 for discussion.
- This includes for
MultiQC v1.7 - 2018-12-21
- BISCUIT
- BISuilfite-seq CUI Toolkit
- Module written by @zwdzwd
- DamageProfiler
- A tool to determine ancient DNA misincorporation rates.
- Module written by @apeltzer
- FLASh
- FLASH (Fast Length Adjustment of SHort reads)
- Module written by @pooranis
- MinIONQC
- QC of reads from ONT long-read sequencing
- Module written by @ManavalanG
- phantompeakqualtools
- A tool for informative enrichment and quality measures for ChIP-seq/DNase-seq/FAIRE-seq/MNase-seq data.
- Module written by @chuan-wang
- Stacks
- A software for analyzing restriction enzyme-based data (e.g. RAD-seq). Support for Stacks >= 2.1 only.
- Module written by @remiolsen
- AdapterRemoval
- Handle error when zero bases are trimmed. See #838.
- Bcl2fastq
- New plot showing the top twenty of undetermined barcodes by lane.
- Informations for R1/R2 are now separated in the General Statistics table.
- SampleID is concatenate with SampleName because in Chromium experiments several sample have the same SampleName.
- deepTools
- New PCA plots from the
plotPCAfunction (written by @chuan-wang) - New fragment size distribution plots from
bamPEFragmentSize --outRawFragmentLengths(written by @chuan-wang) - New correlation heatmaps from the
plotCorrelationfunction (written by @chuan-wang) - New sequence distribution profiles around genes, from the
plotProfilefunction (written by @chuan-wang) - Reordered sections
- New PCA plots from the
- Fastp
- Fixed bug in parsing of empty histogram data. See #845.
- FastQC
- Refactored Per Base Sequence Content plots to show original underlying data, instead of calculating it from the page contents. Now shows original FastQC base-ranges and fixes 100% GC bug in final few pixels. See #812.
- When including a FastQC section multiple times in one report, the summary progress bars now behave as you would expect.
- FastQ Screen
- Don't hide genomes in the simple plot, even if they have zero unique hits. See #829.
- InterOp
- Fixed bug where read counts and base pair yields were not displaying in tables correctly.
- Number formatting for these fields can now be customised in the same way as with other modules, as described in the docs
- Picard
- InsertSizeMetrics: You can now configure to what degree the insert size plot should be smoothed.
- CollectRnaSeqMetrics: Add warning about missing rRNA annotation.
- CollectRnaSeqMetrics: Add chart for counts/percentage of reads mapped to the correct strand.
- Now parses VariantCallingMetrics reports. (Similar to GATK module's VariantEval.)
- phantompeakqualtools
- Properly clean sample names
- Trimmomatic
- Updated Trimmomatic module documentation to be more helpful
- New option to use filenames instead of relying on the command line used. See #864.
- Embed your custom images with a new Custom Content feature! Just add
_mqcto the end of the filename for.png,.jpgor.jpegfiles. - Documentation for Custom Content reordered to make it a little more sane
- You can now add or override any config parameter for any MultiQC plot! See the documentation for more info.
- Allow
table_columns_placementconfig to work with table IDs as well as column namespaces. See #841. - Improved visual spacing between grouped bar plots
- Custom content no longer clobbers
col1_headertable configs - The option
--file-listthat refers to a text file with file paths to analyse will no longer ignore directory paths - Sample name directory prefixes are now added after cleanup.
- If a module is run multiple times in one report, it's CSS and JS files will only be included once (
defaulttemplate)
MultiQC v1.6 - 2018-08-04
Some of these updates are thanks to the efforts of people who attended the NASPM 2018 MultiQC hackathon session. Thanks to everyone who attended!
- fastp
- An ultra-fast all-in-one FASTQ preprocessor (QC, adapters, trimming, filtering, splitting...)
- Module started by @florianduclot and completed by @ewels
- hap.py
- Hap.py is a set of programs based on htslib to benchmark variant calls against gold standard truth datasets
- Module written by @tsnowlan
- Long Ranger
- Works with data from the 10X Genomics Chromium. Performs sample demultiplexing, barcode processing, alignment, quality control, variant calling, phasing, and structural variant calling.
- Module written by @remiolsen
- miRTrace
- A quality control software for small RNA sequencing data.
- Module written by @chuan-wang
- BCFtools
- New plot showing SNP statistics versus quality of call from bcftools stats (@MaxUlysse and @Rotholandus)
- BBMap
- Support added for BBDuk kmer-based adapter/contaminant filtering summary stats (@boulund
- FastQC
- New read count plot, split into unique and duplicate reads if possible.
- Help text added for all sections, mostly copied from the excellent FastQC help.
- Sequence duplication plot rescaled
- FastQ Screen
- Samples in large-sample-number plot are now sorted alphabetically (@hassanfa
- MACS2
- Output is now more tolerant of missing data (no plot if no data)
- Peddy
- Picard
- New submodule to handle
ValidateSamFilereports (@cpavanrun) - WGSMetrics now add the mean and standard-deviation coverage to the general stats table (hidden) (@cpavanrun)
- New submodule to handle
- Preseq
- New config option to plot preseq plots with unique old coverage on the y axis instead of read count
- Code refactoring by @vladsaveliev
- QUAST
- Null values (
-) in reports now handled properly. Bargraphs always shown despite varying thresholds. (@vladsaveliev)
- Null values (
- RNA-SeQC
- Don't create the report section for Gene Body Coverage if no data is given
- Samtools
- Fixed edge case bug where MultiQC could crash if a sample had zero count coverage with idxstats.
- Adds % proper pairs to general stats table
- Skewer
- Read length plot rescaled
- Tophat
- Fixed bug where some samples could be given a blank sample name (@lparsons)
- VerifyBamID
- Change column header help text for contamination to match percentage output (@chapmanb)
- New config option
remove_sectionsto skip specific report sections from modules - Add
path_filters_excludeto exclude certain files when running modules multiple times. You could previously only include certain files. - New
exclude_*keys for file search patterns- Have a subset of patterns to exclude otherwise detected files with, by filename or contents
- Command line options all now use mid-word hyphens (not a mix of hyphens and underscores)
- Old underscore terms still maintained for backwards compatibility
- Flag
--view-tagsnow works without requiring an "analysis directory". - Removed Python dependency for
enum34(@boulund) - Columns can be added to
General Statstable for custom content/module. - New
--ignore-symlinksflag which will ignore symlinked directories and files. - New
--no-megaqc-uploadflag which disables automatically uploading data to MegaQC
- Fix path filters for
top_modules/module_orderconfiguration only selecting if all globs match. It now filters searches that match any glob. - Empty sample names from cleaning are now no longer allowed
- Stop prepend_dirs set in the config from getting clobbered by an unpassed CLI option (@tsnowlan)
- Modules running multiple times now have multiple sets of columns in the General Statistics table again, instead of overwriting one another.
- Prevent tables from clobbering sorted row orders.
- Fix linegraph and scatter plots data conversion (sporadically the incorrect
ymaxwas used to drop data points) (@cpavanrun) - Adjusted behavior of ceiling and floor axis limits
- Adjusted multiple file search patterns to make them more specific
- Prevents the wrong module from accidentally slurping up output from a different tool. By @cpavanrun (see PR #727)
- Fixed broken report bar plots when
-p/--export-plotswas specified (see issue #801)
MultiQC v1.5 - 2018-03-15
- HiCPro - New module!
- HiCPro: Quality controls and processing of Hi-C
- Module written by @nservant,
- DeDup - New module!
- DeDup: Improved Duplicate Removal for merged/collapsed reads in ancient DNA analysis
- Module written by @apeltzer,
- Clip&Merge - New module!
- Clip&Merge: Adapter clipping and read merging for ancient DNA analysis
- Module written by @apeltzer,
- bcl2fastq
- BUSCO
- Fixed configuration bug that made all sample names become
'short'
- Fixed configuration bug that made all sample names become
- Custom Content
- Parsed tables now exported to
multiqc_datafiles
- Parsed tables now exported to
- Cutadapt
- Refactor parsing code to collect all length trimming plots
- FastQC
- Fixed starting y-axis label for GC-content lineplot being incorrect.
- HiCExplorer
- Updated to work with v2.0 release.
- Homer
- Made parsing of
tagInfo.txtfile more resilient to variations in file format so that it works with new versions of Homer. - Kept order of chromosomes in coverage plot consistent.
- Made parsing of
- Peddy
- Switch
Sex errorlogic toCorrect sexfor better highlighting (@aledj2)
- Switch
- Picard
- Updated module and search patterns to recognise new output format from Picard version >= 2.16 and GATK output.
- Qualimap BamQC
- Fixed bug where start of Genome Fraction could have a step if target is 100% covered.
- RNA-SeQC
- Added rRNA alignment stats to summary table @Rolandde
- RSeqC
- Fixed read distribution plot by adding category for
other_intergenic(thanks to @moxgreen) - Fixed a dodgy plot title (Read GC content)
- Fixed read distribution plot by adding category for
- Supernova
- Added support for Supernova 2.0 reports. Fixed a TypeError bug when using txt reports only. Also a bug when parsing empty histogram files.
- Invalid choices for
--moduleor--excludenow list the available modules alphabetically. - Linting now checks for presence in
config.module_orderand tags.
- Excluding modules now works in combination with using module tags.
- Fixed edge-case bug where certain combinations of
output_fn_nameanddata_dir_namecould trigger a crash - Conditional formatting - values are now longer double-labelled
- Made config option
extra_serieswork in scatter plots the same way that it works for line plots - Locked the
matplotlibversion tov2.1.0and below
MultiQC v1.4 - 2018-01-11
A slightly earlier-than-expected release due to a new problem with dependency packages that is breaking MultiQC installations since 2018-01-11.
- Sargasso
- Parses output from Sargasso - a tool to separate mixed-species RNA-seq reads according to their species of origin
- Module written by @hxin
- VerifyBAMID
- Parses output from VerifyBAMID - a tool to detect contamination in BAM files.
- Adds the
CHIPMIXandFREEMIXcolumns to the general statistics table. - Module written by @aledj2
- MACS2
- Updated to work with output from older versions of MACS2 by @avilella
- Peddy
- Add het check plot to suggest potential contamination by @aledj2
- Picard
- Picard HsMetrics
HS_PENALTYplot now has correct axis labels - InsertSizeMetrics switches commas for points if it can't convert floats. Should help some european users.
- Picard HsMetrics
- QoRTs
- Added support for new style of output generated in the v1.3.0 release
- Qualimap
- QUAST
- New option to customise the default display of contig count and length (eg.
bpinstead ofMbp). - See documentation. Written by @ewels and @Cashalow
- New option to customise the default display of contig count and length (eg.
- RSeQC
- Removed normalisation in Junction Saturation plot. Now raw counts instead of % of total junctions.
- Conditional formatting / highlighting of cell contents in tables
- If you want to make values that match a criteria stand out more, you can now write custom rules and formatting instructions for tables.
- For instructions, see the documentation
- New
--lintoption which is strict about best-practices for writing new modules- Useful when writing new modules and code as it throws warnings
- Currently only implemented for bar plots and a few other places. More linting coming soon...
- If MultiQC breaks and shows am error message, it now reports the filename of the last log it found
- Hopefully this will help with debugging / finding dodgy input data
- Addressed new dependency error with conflicting package requirements
- There was a conflict between the
networkx,colormathandspectrareleases. - I previously forced certain software versions to get around this, but
spectrahas now updated with the unfortunate effect of introducing a new dependency clash that halts installation.
- There was a conflict between the
- Fixed newly introduced bug where Custom Content MultiQC config file search patterns had been broken
- Updated pandoc command used in
--pdfto work with new releases of Pandoc - Made config
table_columns_visiblemodule name key matching case insensitive to make less frustrating
MultiQC v1.3 - 2017-11-03
Only for users with custom search patterns for the bowtie or star: you will
need to update your config files - the bowtie search key is now bowtie1,
star_genecounts is now star/genecounts.
For users with custom modules - search patterns must now conform to the search
pattern naming convention: modulename or modulename/anything (the search pattern
string beginning with the name of your module, anything you like after the first /).
- 10X Supernova
- Parses statistics from the de-novo Supernova software.
- Module written by @remiolsen
- BBMap
- deepTools - new module!
- Parse text output from
bamPEFragmentSize,estimateReadFiltering,plotCoverage,plotEnrichment, andplotFingerprint - Module written by @dpryan79
- Parse text output from
- Homer Tag Directory - new submodule!
- Module written by @rdali
- illumina InterOp
- Module to parse metrics from illumina sequencing runs and demultiplexing, generated by the InterOp package
- Module written by @matthdsm
- RSEM - new module!
- Parse
.cntfile comming from rsem-calculate-expression and plot read repartitions (Unalignable, Unique, Multi ...) - Module written by @noirot
- Parse
- HiCExplorer
- New module to parse the log files of
hicBuildMatrix. - Module written by @joachimwolff
- New module to parse the log files of
- AfterQC
- Handle new output format where JSON summary key changed names.
- bcl2fastq
- Clusters per sample plot now has tab where counts are categoried by lane.
- GATK
- New submodule to handle Base Recalibrator stats, written by @winni2k
- HiSAT2
- Fixed bug where plot title was incorrect if both SE and PE bargraphs were in one report
- Picard HsMetrics
- Parsing code can now handle commas for decimal places
- Preseq
- Updated odd file-search pattern that limited input files to 500kb
- QoRTs
- Added new plots, new helptext and updated the module to produce a lot more output.
- Qualimap BamQC
- Fixed edge-case bug where the refactored coverage plot code could raise an error from the
rangecall.
- Fixed edge-case bug where the refactored coverage plot code could raise an error from the
- Documentation and link fixes for Slamdunk, GATK, bcl2fastq, Adapter Removal, FastQC and main docs
- Many of these spotted and fixed by @juliangehring
- Went through all modules and standardised plot titles
- All plots should now have a title with the format Module name: Plot name
- New MultiQC docker image
- Ready to use docker image now available at https://hub.docker.com/r/MultiQC/MultiQC/ (200 MB)
- Uses automated builds - pull
:latestto get the development version, future releases will have stable tags. - Written by @MaxUlysse
- New
module_orderconfig options allow modules to be run multiple times- Filters mean that a module can be run twice with different sets of files (eg. before and after trimming)
- Custom module config parameters can be passed to module for each run
- File search refactored to only search for running modules
- Makes search much faster when running with lots of files and limited modules
- For example, if using
-m starto only use the STAR module, all other file searches now skipped
- File search now warns if an unrecognised search type is given
- MultiQC now saves nearly all parsed data to a structured output file by default
- See
multiqc_data/multiqc_data.json - This can be turned off by setting
config.data_dump_file: false
- See
- Verbose logging when no log files found standardised. Less duplication in code and logs easier to read!
- New documentation section describing how to use MultiQC with Galaxy
- Using
shared_key: 'read_counts'in table header configs now applies relevant defaults
- Installation problem caused by changes in upstream dependencies solved by stricter installation requirements
- Minor
default_devdirectory creation bug squashed - Don't prepend the directory separator (
|) to sample names with-dwhen there are no subdirs yPlotLinesnow works even if you don't setwidth
MultiQC v1.2 - 2017-08-16
We had a fantastic group effort on MultiQC at the 2017 BOSC CodeFest. Many thanks to those involved!
- AfterQC - New module!
- Added parsing of the AfterQC json file data, with a plot of filtered reads.
- Work by @raonyguimaraes
- bcl2fastq
- leeHom
- leeHom is a program for the Bayesian reconstruction of ancient DNA
- VCFTools
- Added initial support for VCFTools
relatedness2 - Added support for VCFTools
TsTv-by-countTsTv-by-qualTsTv-summary - Module written by @mwhamgenomics
- Added initial support for VCFTools
- FastQ Screen
- Gracefully handle missing data from very old FastQ Screen versions.
- RNA-SeQC
- Add new transcript-associated reads plot.
- Picard
- New submodule to handle output from
TargetedPcrMetrics
- New submodule to handle output from
- Prokka
- Added parsing of the
# CRISPR arraysdata from Prokka when available (@asetGem)
- Added parsing of the
- Qualimap
- Some code refactoring to radically improve performance and run times, especially with high coverage datasets.
- Fixed bug where Cumulative coverage genome fraction plot could be truncated.
- New module help text
- Lots of additional help text was written to make MultiQC report plots easier to interpret.
- Updated modules:
- Bowtie
- Bowtie 2
- Prokka
- Qualimap
- SnpEff
- Elite team of help-writers:
- New config option
section_commentsallows you to add custom comments above specific sections in the report - New
--tagsand--view_tagscommand line options- Modules can now be given tags (keywords) and filtered by those. So running
--tags RNAwill only run MultiQC modules related to RNA analysis. - Work by @Hammarn
- Modules can now be given tags (keywords) and filtered by those. So running
- Back-end configuration options to specify the order of table columns
- Modules and user configs can set priorities for columns to customise where they are displayed
- Work by @tbooth
- Added framework for proper unit testing
- Previous start on unit tests tidied up, new blank template and tests for the
clean_sample_namefunctionality. - Added to Travis and Appveyor for continuous integration testing.
- Work by @tbooth
- Previous start on unit tests tidied up, new blank template and tests for the
- Bug fixes and refactoring of report configuration saving / loading
- Discovered and fixed a bug where a report config could only be loaded once
- Work by @DennisSchwartz
- Table column row headers (sample names) can now be numeric-only.
- Work by @iimog
- Improved sample name cleaning functionality
- Added option
regex_keepto clean filenames by keeping the matching part of a pattern - Work by @robinandeer
- Added option
- Handle error when invalid regexes are given in reports
- Now have a nice toast error warning you and the invalid regexes are highlighted
- Previously this just crashed the whole report without any warning
- Work by @robinandeer
- Command line option
--dirs-depthnow sets-dtoTrue(so now works even if-disn't also specified). - New config option
config.data_dump_fileto export as much data as possible tomultiqc_data/multiqc_data.json - New code to send exported JSON data to a a web server
- This is in preparation for the upcoming MegaQC project. Stay tuned!
- Specifying multiple config files with
-c/--confignow works as expected- Previously this would only read the last specified
- Fixed table rendering bug that affected Chrome v60 and IE7-11
- Table cell background bars weren't showing up. Updated CSS to get around this rendering error.
- HTML ID cleanup now properly cleans strings so that they work with jQuery as expected.
- Made bar graph sample highlighting work properly again
- Config
custom_logopaths can now be relative to the config file (or absolute as before) - Report doesn't keep annoyingly telling you that toolbox changes haven't been applied
- Now uses more subtle toasts and only when you close the toolbox (not every click).
- Switching report toolbox options to regex mode now enables the Apply button as it should.
- Sorting table columns with certain suffixes (eg.
13X) no works properly (numerically) - Fixed minor bug in line plot data smoothing (now works with unsorted keys)
MultiQC v1.1 - 2017-07-18
- BioBloom Tools
- Create Bloom filters for a given reference and then to categorize sequences
- Conpair
- Concordance and contamination estimator for tumor–normal pairs
- Disambiguate
- Bargraph displaying the percentage of reads aligning to two different reference genomes.
- Flexbar
- Flexbar is a tool for flexible barcode and adapter removal.
- HISAT2
- New module for the HISAT2 aligner.
- Made possible by updates to HISAT2 logging by @infphilo (requires
--new-summaryHISAT2 flag).
- HOMER
- Support for summary statistics from the
findPeakstool.
- Support for summary statistics from the
- Jellyfish
- Histograms to estimate library complexity and coverage from k-mer content.
- Module written by @vezzi
- MACS2
- Summary of redundant rate from MACS2 peak calling.
- QoRTs
- QoRTs is toolkit for analysis, QC and data management of RNA-Seq datasets.
- THetA2
- THeTA2 (Tumor Heterogeneity Analysis) estimates tumour purity and clonal / subclonal copy number.
- BCFtools
- Option to collapse complementary changes in substitutions plot, useful for non-strand specific experiments (thanks to @vladsaveliev)
- Bismark
- M-Bias plots no longer show read 2 for single-end data.
- Custom Content
- New option to print raw HTML content to the report.
- FastQ Screen
- Fixed edge-case bug where many-sample plot broke if total number of reads was less than the subsample number.
- Fixed incorrect logic of config option
fastqscreen_simpleplot(thanks to @daler) - Organisms now alphabetically sorted in fancy plot so that order is nonrandom (thanks to @daler)
- Fixed bug where
%No Hitswas missed in logs from recent versions of FastQ Screen.
- HTSeq Counts
- Fixed but so that module still works when
--additional-attris specified in v0.8 HTSeq above (thanks to @nalcala)
- Fixed but so that module still works when
- Picard
- CollectInsertSize: Fixed bug that could make the General Statistics Median Insert Size value incorrect.
- Fixed error in sample name regex that left trailing
]characters and was generally broken (thanks to @jyh1 for spotting this)
- Preseq
- Improved plots display (thanks to @vladsaveliev)
- Qualimap
- Only calculate bases over target coverage for values in General Statistics. Should give a speed increase for very high coverage datasets.
- QUAST
- Module is now compatible with runs from MetaQUAST (thanks to @vladsaveliev)
- RSeQC
- Changed default order of sections
- Added config option to reorder and hide module report sections
- If a report already exists, execution is no longer halted.
_1is appended to the filename, iterating if this also exists.-f/--forcestill overwrites existing reports as before- Feature written by @Hammarn
- New ability to run modules multiple times in a single report
- Each run can be given different configuration options, including filters for input files
- For example, have FastQC after trimming as well as FastQC before trimming.
- See the relevant documentation for more instructions.
- New option to customise the order of report sections
- This is in addition / alternative to changing the order of module execution
- Allows one module to have sections in multiple places (eg. Custom Content)
- Tables have new column options
floor,ceilingandminRange. - Reports show warning if JavaScript is disabled
- Config option
custom_logonow works with file paths relative to config file directory and cwd.
- Table headers now sort columns again after scrolling the table
- Fixed buggy table header tooltips
- Base
clean_s_namefunction now strips excess whitespace. - Line graphs don't smooth lines if not needed (number of points < maximum number allowed)
- PDF output now respects custom output directory.
MultiQC v1.0 - 2017-05-17
Version 1.0! This release has been a long time coming and brings with it some fairly major improvements in speed, report filesize and report performance. There's also a bunch of new modules, more options, features and a whole lot of bug fixes.
The version number is being bumped up to 1.0 for a couple of reasons:
- MultiQC is now (hopefully) relatively stable. A number of facilities and users are now using it in a production setting and it's published. It feels like it probably deserves v1 status now somehow.
- This update brings some fairly major changes which will break backwards compatibility for plugins. As such, semantic versioning suggests a change in major version number.
For most people, you shouldn't have any problems upgrading. There are two scenarios where you may need to make changes with this update:
Search patterns have been flattened and may no longer have arbitrary depth. For example, you may need to change the following:
fastqc:
data:
fn: "fastqc_data.txt"
zip:
fn: "*_fastqc.zip"to this:
fastqc/data:
fn: "fastqc_data.txt"
fastqc/zip:
fn: "*_fastqc.zip"See the documentation for instructions on how to write the new file search syntax.
See search_patterns.yaml for the new module search keys
and more examples.
To see what changes need to applied to your custom plugin code, please see the MultiQC docs.
- Adapter Removal
- AdapterRemoval v2 - rapid adapter trimming, identification, and read merging
- BUSCO
- New module for the
BUSCO v2tool, used for assessing genome assembly and annotation completeness.
- New module for the
- Cluster Flow
- Cluster Flow is a workflow tool for bioinformatics pipelines. The new module parses executed tool commands.
- RNA-SeQC
- New module to parse output from RNA-SeQC, a java program which computes a series of quality control metrics for RNA-seq data.
- goleft indexcov
- goleft indexcov uses the PED and ROC data files to create diagnostic plots of coverage per sample, helping to identify sample gender and coverage issues.
- Thanks to @chapmanb and @brentp
- SortMeRNA
- New module for
SortMeRNA, commonly used for removing rRNA contamination from datasets. - Written by @bschiffthaler
- New module for
- Bcftools
- Fixed bug with display of indels when only one sample
- Cutadapt
- Now takes the filename if the sample name is
-(stdin). Thanks to @tdido
- Now takes the filename if the sample name is
- FastQC
- Data for the Sequence content plot can now be downloaded from reports as a JSON file.
- FastQ Screen
- Rewritten plotting method for high sample numbers plot (~ > 20 samples)
- Now shows counts for single-species hits and bins all multi-species hits
- Allows plot to show proper percentage view for each sample, much easier to interpret.
- HTSeq
- Fix bug where header lines caused module to crash
- Picard
- New
RrbsSummaryMetricsSubmodule! - New
WgsMetricsSubmodule! CollectGcBiasMetricsmodule now prints summary statistics tomultiqc_dataif found. Thanks to @ahvigil
- New
- Preseq
- Now trims the x axis to the point that meets 90% of
min(unique molecules). Hopefully prevents ridiculous x axes without sacrificing too much useful information. - Allows to show estimated depth of coverage instead of less informative molecule counts (see details).
- Plots dots with externally calculated real read counts (see details).
- Now trims the x axis to the point that meets 90% of
- Qualimap
- RNASeq Transcript Profile now has correct axis units. Thanks to @roryk
- BamQC module now doesn't crash if reports don't have genome gc distributions
- RSeQC
- Fixed Python3 error in Junction Saturation code
- Fixed JS error for Junction Saturation that made the single-sample combined plot only show All Junctions
- Change in module structure and import statements (see details).
- Module file search has been rewritten (see above changes to configs)
- Significant improvement in search speed (test dataset runs in approximately half the time)
- More options for modules to find their logs, eg. filename and contents matching regexes (see the docs)
- Report plot data is now compressed, significantly reducing report filesizes.
- New
--ignore-samplesoption to skip samples based on parsed sample name- Alternative to filtering by input filename, which doesn't always work
- Also can use config vars
sample_names_ignore(glob patterns) andsample_names_ignore_re(regex patterns).
- New
--sample-namescommand line option to give file with alternative sample names- Allows one-click batch renaming in reports
- New
--cl_configoption to supply MultiQC config YAML directly on the command line. - New config option to change numeric multiplier in General Stats
- For example, if reports have few reads, can show
Thousands of Readsinstead ofMillions of Reads - Set config options
read_count_multiplier,read_count_prefixandread_count_desc
- For example, if reports have few reads, can show
- Config options
decimalPoint_formatandthousandsSep_formatnow apply to tables as well as plots- By default, thosands will now be separated with a space and
.used for decimal places.
- By default, thosands will now be separated with a space and
- Tables now have a maximum-height by default and scroll within this.
- Speeds up report rendering in the web browser and makes report less stupidly long with lots of samples
- Button beneath table toggles full length if you want a zoomed-out view
- Refactored and removed previous code to make the table header "float"
- Set
config.collapse_tablestoFalseto disable table maximum-heights
- Bar graphs and heatmaps can now be zoomed in on
- Interactive plots sometimes hide labels due to lack of space. These can now be zoomed in on to see specific samples in more detail.
- Report plots now load sequentially instead of all at once
- Prevents the browser from locking up when large reports load
- Report plot and section HTML IDs are now sanitised and checked for duplicates
- New template available (called sections) which has faster loading
- Only shows results from one module at a time
- Makes big reports load in the browser much more quickly, but requires more clicking
- Try it out by specifying
-t sections
- Module sections tidied and refactored
- New helper function
self.add_section() - Sections hidden in nav if no title (no more need for the hacky
self.intro +=) - Content broken into
description,helpandplot, with automatic formatting - Empty module sections are now skipped in reports. No need to check if a plot function returns
None! - Changes should be backwards-compatible
- New helper function
- Report plot data export code refactored
- Now doesn't export hidden samples (uses HighCharts export-csv plugin)
- Handle error when
gitisn't installed on the system. - Refactored colouring of table cells
- Docs updates (thanks to @varemo)
- Previously hidden log file
.multiqc.logrenamed tomultiqc.loginmultiqc_data - Added option to load MultiQC config file from a path specified in the environment variable
MULTIQC_CONFIG_PATH - New table configuration options
sortRows: Falseprevents table rows from being sorted alphabeticallycol1_headerallows the default first column header to be changed from "Sample Name"
- Tables no longer show Configure Columns and Plot buttons if they only have a single column
- Custom content updates
- New
custom_content/orderconfig option to specify order of Custom Content sections - Tables now use the header for the first column instead of always having
Sample Name - JSON + YAML tables now remember order of table columns
- Many minor bugfixes
- New
- Line graphs and scatter graphs axis limits
- If limits are specified, data exceeding this is no longer saved in report
- Visually identical, but can make report file sizes considerable smaller in some cases
- Creating multiple plots without a config dict now works (previously just gave grey boxes in report)
- All changes are now tested on a Windows system, using AppVeyor
- Fixed rare error where some reports could get empty General Statistics tables when no data present.
- Fixed minor bug where config option
force: truedidn't work. Now you don't have to always specify-f!
MultiQC v0.9 - 2016-12-21
A major new feature is released in v0.9 - support for custom content. This means that MultiQC can now easily include output from custom scripts within reports without the need for a new module or plugin. For more information, please see the MultiQC documentation.
- HTSeq
- New module for the
htseq-counttool, often used in RNA-seq analysis.
- New module for the
- Prokka
- Prokka is a software tool for the rapid annotation of prokaryotic genomes.
- Slamdunk
- Slamdunk is a software tool to analyze SLAMSeq data.
- Peddy
- Peddy calculates genotype :: pedigree correspondence checks, ancestry checks and sex checks using VCF files.
- Cutadapt
- Fixed bug in General Stats table number for old versions of cutadapt (pre v1.7)
- Added support for really old cutadapt logs (eg. v.1.2)
- FastQC
- New plot showing total overrepresented sequence percentages.
- New option to parse a file containing a theoretical GC curve to display in the background.
- Human & Mouse Genome / Transcriptome curves bundled, or make your own using fastqcTheoreticalGC. See the MultiQC docs for more information.
- featureCounts
- Added parsing checks and catch failures for when non-featureCounts files are picked up by accident
- GATK
- Fixed logger error in VariantEval module.
- Picard
- Fixed missing sample overwriting bug in
RnaSeqMetrics - New feature to customise coverage shown from
HsMetricsin General Statistics table see the docs for info). - Fixed compatibility problem with output from
CollectMultipleMetricsforCollectAlignmentSummaryMetrics
- Fixed missing sample overwriting bug in
- Preseq
- Module now recognises output from
c_curvemode.
- Module now recognises output from
- RSeQC
- Made the gene body coverage plot show the percentage view by default
- Made gene body coverage properly handle sample names
- Samtools
- New module to show duplicate stats from
rmduplogs - Fixed a couple of niggles in the idxstats plot
- New module to show duplicate stats from
- SnpEff
- Fixed swapped axis labels in the Variant Quality plot
- STAR
- Fixed crash when there are 0 unmapped reads.
- Sample name now taken from the directory name if no file prefix found.
- Qualimap BamQC
- Add a line for pre-calculated reference genome GC content
- Plot cumulative coverage for values above 50x, align with the coverage histogram.
- New ability to customise coverage thresholds shown in General Statistics table (see the docs for info).
- Support for custom content (see top of release notes).
- New ninja report tool: make scatter plots of any two table columns!
- Plot data now saved in
multiqc_datawhen 'flat' image plots are created- Allows you easily re-plot the data (eg. in Excel) for further downstream investigation
- Added 'Apply' button to Highlight / Rename / Hide.
- These tools can become slow with large reports. This means that you can enter several things without having to wait for the report to replot each change.
- Report heatmaps can now be sorted by highlight
- New config options
decimalPoint_formatandthousandsSep_format- Allows you to change the default
1 234.56number formatting for plots.
- Allows you to change the default
- New config option
top_modulesallows you to specify modules that should come at the top of the report - Fixed bar plot bug where missing categories could shift data between samples
- Report title now printed in the side navigation
- Missing plot IDs added for easier plot exporting
- Stopped giving warnings about skipping directories (now a debug message)
- Added warnings in report about missing functionality for flat plots (exporting and toolbox)
- Export button has contextual text for images / data
- Fixed a bug where user config files were loaded twice
- Fixed bug where module order was random if
--moduleor--excludewas used. - Refactored code so that the order of modules can be changed in the user config
- Beefed up code + docs in scatter plots back end and multiple bar plots.
- Fixed a few back end nasties for Tables
- Shared-key columns are no longer forced to share colour schemes
- Fixed bug in lambda modified values when format string breaks
- Supplying just data with no header information now works as advertised
- Improvements to back end code for bar plots
- New
tt_decimalsandtt_suffixoptions for bar plots - Bar plots now support
yCeiling,yFloorandyMinRange, as with line plots. - New option
hide_zero_cats:Falseto force legends to be shown even when all data is 0
- New
- General Stats Showing x of y columns count is fixed on page load.
- Big code whitespace cleanup
MultiQC v0.8 - 2016-09-26
- GATK
- Added support for VariantEval reports, only parsing a little of the information in there so far, but it's a start.
- Module originally written by @robinandeer at the OBF Codefest, finished off by @ewels
- Bcftools
- QUAST
- QUAST is a tool for assessing de novo assemblies against reference genomes.
- Bismark now supports reports from
bam2nuc, giving Cytosine coverage in General Stats. - Bowtie1
- Updated to try to find bowtie command before log, handle multiple logs in one file. Same as bowtie2.
- FastQC
- Sample pass/warn/fail lists now display properly even with large numbers of samples
- Sequence content heatmap display is better with many samples
- Kallisto
- Now supports logs from SE data.
- Picard
BaseDistributionByCycle- new submodule! Written by @mlusignanRnaSeqMetrics- new submodule! This one by @ewels ;)AlignmentSummaryMetrics- another new submodule!- Fixed truncated files crash bug for Python 3 (#306)
- Qualimap RNASeqQC
- Fixed parsing bug affecting counts in Genomic Origin plot.
- Module now works with European style thousand separators (
1.234,56instead of1,234.56)
- RSeQC
infer_experiment- new submodule! Written by @Hammarn
- Samtools
statssubmodule now has separate bar graph showing alignment scoresflagstat- new submodule! Written by @HLWienckoidxstats- new submodule! This one by @ewels again
- New
--export/-poption to generate static images plot inmultiqc_plots(.png,.svgand.pdf)- Configurable with
export_plots,plots_dir_nameandexport_plot_formatsconfig options --flatoption no longer saves plots inmultiqc_data/multiqc_plots
- Configurable with
- New
--comment/-bflag to add a comment to the top of reports. - New
--dirs-depth/-ddflag to specify how many directories to prepend with--dirs/-d- Specifying a postive number will take that many directories from the end of the path
- A negative number will take directories from the start of the path.
- Directory paths now appended before cleaning, so
fn_clean_extswill now affect these names. - New
custom_logoattributes to add your own logo to reports. - New
report_header_infoconfig option to add arbitrary information to the top of reports. - New
--pdfoption to create a PDF report- Depends on Pandoc being installed and is in a beta-stage currently.
- Note that specifying this will make MultiQC use the
simpletemplate, giving a HTML report with much reduced functionality.
- New
fn_clean_sample_namesconfig option to turn off sample name cleaning- This will print the full filename for samples. Less pretty reports and rows on the General Statistics table won't line up, but can prevent overwriting.
- Table header defaults can now be set easily
- General Statistics table now hidden if empty.
- Some new defaults in the sample name cleaning
- Updated the
simpletemplate.- Now has no toolbox or nav, no JavaScript and is better suited for printing / PDFs.
- New
config.simple_outputconfig flag so code knows when we're trying to avoid JS.
- Fixed some bugs with config settings (eg. template) being overwritten.
- NFS log file deletion bug fixed by @brainstorm (#265)
- Fixed bug in
--ignorebehaviour with directory names. - Fixed nasty bug in beeswarm dot plots where sample names were mixed up (#278)
- Beeswarm header text is now more informative (sample count with more info on a tooltip)
- Beeswarm plots now work when reports have > 1000 samples
- Fixed some buggy behaviour in saving / loading report highlighting + renaming configs (#354)
Many thanks to those at the OpenBio Codefest 2016 who worked on MultiQC projects.
MultiQC v0.7 - 2016-07-04
- Kallisto - new module!
- Picard
- Code refactored to make maintenance and additions easier.
- Big update to
HsMetricsparsing - more results shown in report, new plots (by @lpantano) - Updated
InsertSizeMetricsto understand logs generated byCollectMultipleMetrics(#215) - Newlines in picard output. Fixed by @dakl
- Samtools
- Code refactored
- Rewrote the
samtools statscode to display more stats in report with a beeswarm plot.
- Qualimap
- Rewritten to use latest methods and fix bugs.
- Added Percentage Aligned column to general stats for
BamQCmodule. - Extra table thresholds added by @avilella (hidden by default)
- General Statistics
- Some tweaks to the display defaults (FastQC, Bismark, Qualimap, SnpEff)
- Now possible to skip the General Statistics section of the report with
--exclude general_stats
- Cutadapt module updated to recognise logs from old versions of cutadapt (
<= v1.6) - Trimmomatic
- Now handles
,decimal places in percentage values. - Can cope with line breaks in log files (see issue #212)
- Now handles
- FastQC refactored
- Now skips zip files if the sample name has already been found. Speeds up MultiQC execution.
- Code cleaned up. Parsing and data-structures standardised.
- New popovers on Pass / Warn / Fail status bars showing sample names. Fast highlighting and hiding.
- New column in General Stats (hidden by default) showing percentage of FastQC modules that failed.
- SnpEff
- Search pattern now more generic, should match reports from others.
- Counts by Effect plot removed (had hundreds of categories, was fairly unusable).
KeyErrorbug fixed.
- Samblaster now gets sample name from
IDinstead ofSM(@dakl) - Bowtie 2
- Now parses overall alignment rate as intended.
- Now depends on even less log contents to work with more inputs.
- MethylQA now handles variable spacing in logs
- featureCounts now splits columns on tabs instead of whitespace, can handle filenames with spaces
- Galaxy: MultiQC now available in Galax! Work by @devengineson / @yvanlebras / @cmonjeau
- See it in the Galaxy Toolshed
- Heatmap: New plot type!
- Scatter Plot: New plot type!
- Download raw data behind plots in reports! Available in the Export toolbox.
- Choose from tab-separated, comma-separated and the complete JSON.
- Table columns can be hidden on page load (shown through Configure Columns)
- Defaults are configurable using the
table_columns_visibleconfig option.
- Defaults are configurable using the
- Beeswarm plot: Added missing rename / highlight / hiding functionality.
- New
-l/--file-listoption: specify a file containing a list of files to search. - Updated HighCharts to v4.2.5. Added option to export to JPEG.
- Can now cancel execution with a single
ctrl+crather than having to button mash - More granular control of skipping files during scan (filename, dirname, path matching)
- Fixed
--excludeso that it works with directories as well as files
- Fixed
- New Clear button in toolbox to bulk remove highlighting / renaming / hiding filters.
- Improved documentation about behaviour for large sample numbers.
- Handle YAML parsing errors for the config file more gracefully
- Removed empty columns from tables again
- Fixed bug in changing module search patterns, reported by @lweasel
- Added timeout parameter to version check to prevent hang on systems with long defaults
- Fixed table display bug in Firefox
- Fixed bug related to order in which config files are loaded
- Fixed bug that broke the "Show only" toolbox feature with multiple names.
- Numerous other small bugs.
MultiQC v0.6 - 2016-04-29
- New Salmon module.
- New Trimmomatic module.
- New Bamtools stats module.
- New beeswarm plot type. General Stats table replaced with this when many samples in report.
- New RSeQC module: Actually a suite of 8 new modules supporting various outputs from RSeQC
- Rewrote bowtie2 module: Now better at parsing logs and tries to scrape input from wrapper logs.
- Made cutadapt show counts by default instead of obs/exp
- Added percentage view to Picard insert size plot
- Dynamic plots now update their labels properly when changing datasets and to percentages
- Config files now loaded from working directory if present
- Started new docs describing how each module works
- Refactored featureCounts module. Now handles summaries describing multiple samples.
- Stopped using so many hidden files.
.multiqc.lognow calledmultiqc.log - New
-c/--configcommand line option to specify a MultiQC configuration file - Can now load run-specific config files called
multiqc_config.yamlin working directory - Large code refactoring - moved plotting code out of
BaseModuleand into newmultiqc.plotssubmodules - Generalised code used to generate the General Stats table so that it can be used by modules
- Removed interactive report tour, replaced with a link to a youtube tutorial
- Made it possible to permanently hide the blue welcome message for all future reports
- New option to smooth data for line plots. Avoids mega-huge plots. Applied to SnpEff, RSeQC, Picard.
Bugfixes:
- Qualimap handles infinity symbol (thanks @chapmanb )
- Made SnpEff less fussy about required fields for making plots
- UTF-8 file paths handled properly in Py2.7+
- Extending two config variables wasn't working. Now fixed.
- Dragging the height bar of plots now works again.
- Plots now properly change y axis limits and labels when changing datasets
- Flat plots now have correct path in
default_devtemplate
MultiQC v0.5 - 2016-03-29
- New Skewer module, written by @dakl
- New Samblaster module, written by @dakl
- New Samtools stats module, written by @lpantano
- New HiCUP module
- New SnpEff module
- New methylQA module
- New "Flat" image plots, rendered at run time with MatPlotLib
- By default, will use image plots if > 50 samples (set in config as
plots_flat_numseries) - Means that very large numbers of samples can be viewed in reports. eg. single cell data.
- Templates can now specify their own plotting functions
- Use
--flatand--interactiveto override this behaviour
- By default, will use image plots if > 50 samples (set in config as
- MultiQC added to
bioconda(with help from @dakl) - New plugin hook:
config_loaded - Plugins can now add new command line options (thanks to @robinandeer)
- Changed default data directory name from
multiqc_report_datatomultiqc_data - Removed support for depreciated MultiQC_OSXApp
- Updated logging so that a verbose
multiqc_data/.multiqc.logfile is always written - Now logs more stuff in verbose mode - command used, user configs and so on.
- Added a call to multiqc.info to check for new versions. Disable with config
no_version_check - Removed general stats manual row sorting.
- Made filename matching use glob unix style filename match patterns
- Everything (including the data directory) is now created in a temporary directory and moved when MultiQC is complete.
- A handful of performance updates for large analysis directories
MultiQC v0.4 - 2016-02-16
- New
multiqc_sources.txtwhich identifies the paths used to collect all report data for each sample - Export parsed data as tab-delimited text,
JSONorYAMLusing the new-k/--data-formatcommand line option - Updated HighCharts from
v4.2.2tov4.2.3, fixes tooltip hover bug. - Nicer export button. Now tied to the export toolbox, hopefully more intuitive.
- FastQC: Per base sequence content heatmap can now be clicked to show line graph for single sample
- FastQC: No longer show adapter contamination datasets with
<= 0.1%contamination. - Picard: Added support for
CollectOxoGMetricsreports. - Changed command line option
--nameto--filename --namealso used for filename if--filenamenot specified.- Hide samples toolbox now has switch to show only matching samples
- New regex help box with examples added to report
- New button to copy general stats table to the clipboard
- General Stats table 'floating' header now sorts properly when scrolling
- Bugfix: MultiQC default_dev template now copies module assets properly
- Bufgix: General Stats table floating header now resizes properly when page width changes
MultiQC v0.3.2 - 2016-02-08
- All modules now load their log file search parameters from a config
file, allowing you to overwrite them using your user config file
- This is useful if your analysis pipeline renames program outputs
- New Picard (sub)modules - Insert Size, GC Bias & HsMetrics
- New Qualimap (sub)module - RNA-Seq QC
- Made Picard MarkDups show percent by default instead of counts
- Added M-Bias plot to Bismark
- New option to stream report HTML to
stdout - Files can now be specified as well as directories
- New options to specify whether the parsed data directory should be created
- command line flags:
--data/--no-data - config option name:
make_data_dir
- command line flags:
- Fixed bug with incorrect path to installation dir config YAML file
- New toolbox drawer for bulk-exporting graph images
- Report side navigation can now be hidden to maximise horizontal space
- Mobile styling improved for narrow screen
- More vibrant colours in the general stats table
- General stats table numbers now left aligned
- Settings now saved and loaded to named localstorage locations
- Simplified interface - no longer global / single report saving
- Removed static file config. Solves JS error, no-one was doing this since we have standalone reports anyway.
- Added support for Python 3.5
- Fixed bug with module specific CSS / JS includes in some templates
- Made the 'ignore files' config use unix style file pattern matching
- Fixed some bugs in the FastQ Screen module
- Fixed some bugs in the FastQC module
- Fixed occasional general stats table bug
- Table sorting on sample names now works after renaming
- Bismark module restructure
- Each report type now handled independently (alignment / dedup / meth extraction)
- M-Bias plot now shows R1 and R2
- FastQC GC content plot now has option for counts or percentages
- Allows comparison between samples with very different read counts
- Bugfix for reports javascript
- Caused by updated to remotely loaded HighCharts export script
- Export script now bundled with multiqc, so does not depend on internet connection
- Other JS errors fixed in this work
- Bugfix for older FastQC reports - handle old style sequence dup data
- Bugfix for varying Tophat alignment report formats
- Bugfix for Qualimap RNA Seq reports with paired end data
MultiQC v0.3.1 - 2015-11-04
- Hotfix patch to fix broken FastQC module (wasn't finding
.zipfiles properly) - General Stats table colours now flat. Should improve browser speed.
- Empty rows now hidden if appear due to column removal in general stats
- FastQC Kmer plot removed until we have something better to show.
MultiQC v0.3 - 2015-11-04
- Lots of lovely new documentation!
- Child templates - easily customise specific parts of the default report template
- Plugin hooks - allow other tools to execute custom code during MultiQC execution
- New Preseq module
- New design for general statistics table (snazzy new background bars)
- Further development of toolbox
- New button to clear all filters
- Warnings when samples are hidden, plus empty plots and table cols are hidden
- Active toolbar tab buttons are highlighted
- Lots of refactoring by @moonso to please the Pythonic gods
- Switched to click instead of argparse to handle command line arguments
- Code generally conforms to best practices better now.
- Now able to supply multiple directories to search for reports
- Logging output improved (now controlled by
-qand-vfor quiet and verbose) - More HTML output dealt with by the base module, less left to the modules
- Module introduction text
- General statistics table now much easier to add to (new helper functions)
- Images, CSS and Javascript now included in HTML, meaning that there is a single report file to make sharing easier
- More accessible scrolling in the report - styled scrollbars and 'to top' button.
- Modules and templates now use setuptools entry points, facilitating plugins by other packages. Allows niche extensions whilst keeping the core codebase clean.
- The general stats table now has a sticky header row when scrolling, thanks to some new javascript wizardry...
- General stats columns can have a shared key which allows common colour schemes and data ranges. For instance, all columns describing a read count will now share their scale across modules.
- General stats columns can be hidden and reordered with a new modal window.
- Plotting code refactored, reports with many samples (>50 by default) don't automatically render to avoid freezing the browser.
- Plots with highlighted and renamed samples now honour this when exporting to different file types.
MultiQC v0.2 - 2015-09-18
- Code restructuring for nearly all modules. Common base module
functions now handle many more functions (plots, config, file import)
- See the contributing notes for instructions on how to use these new helpers to make your own module
- New report toolbox - sample highlighting, renaming, hiding
- Config is autosaved by default, can also export to a file for sharing
- Interactive tour to help users find their way around
- New Tophat, Bowtie 2 and QualiMap modules
- Thanks to @guillermo-carrasco for the QualiMap module
- Bowtie module now works
- New command line parameter
-dprefixes sample names with the directory that they were found in. Allows duplicate filenames without being overwritten. - Introduction walkthrough helps show what can be done in the report
- Now compatible with both Python 2 and Python 3
- Software version number now printed on command line properly, and in reports.
- Bugfix: FastQC doesn't break when only one report found
- Bugfix: FastQC seq content heatmap highlighting
- Many, many small bugfixes
MultiQC v0.1 - 2015-09-01
- The first public release of MultiQC, after a month of development. Basic structure in place and modules for FastQC, FastQ Screen, Cutadapt, Bismark, STAR, Bowtie, Subread featureCounts and Picard MarkDuplicates. Approaching stability, though still under fairly heavy development.